
Rad52 Oligomeric N-Terminal Domain Stabilizes Rad51 Nucleoprotein Filaments and Contributes to Their Protection against Srs2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. S. Cerevisiae Strains
2.2. Directed Mutagenesis
2.3. Sequence Alignment
2.4. Irradiation and Measurement of Recombination Rates
2.5. Survival Following DNA DSB Formation
2.6. Structure
2.7. Cycloheximide Expression Shut-Off Experiment
2.8. Y2H Assay
2.9. Co-Immunoprecipitation
2.10. ChIP Experiments and Quantitative PCR Analyses
2.11. Protein Purification
2.12. Electron Microscopy Analysis
2.13. Electrophoretic Mobility Shift Assay
2.14. DNA Annealing
2.15. DNA Strand Exchange Reaction
2.16. Challenging Rad51 Filaments with Excess Amounts of DNA
3. Results
3.1. Mutations Affecting Conserved Residues of the N-Terminal Domain of Rad52 Suppress Rad51 Filament Toxicity in Srs2-Deficient Cells
3.2. N-Terminal Rad52 Mutations That Suppress Rad51 Filament Toxicity Are Proficient for HR in Srs2-Deficient Cells
3.3. Gene Conversion and SSA Still Occur in the Rad52 N-Terminal Mutants
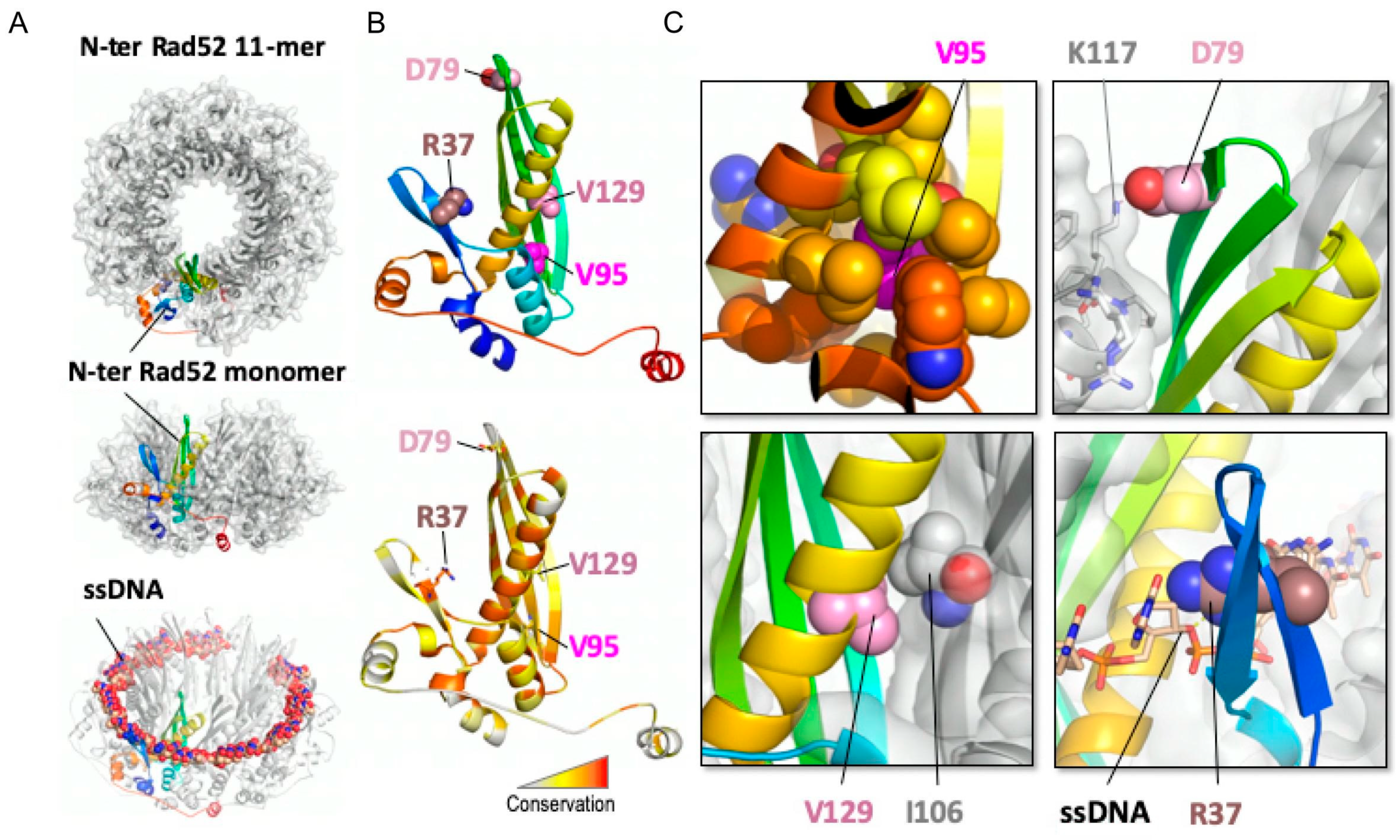
3.4. Structural Analysis of the Rad52 N-Terminal Domain Suggests That Rad52-V95I, Rad52-V129A and Rad52-D79N Affect Rad52 Oligomerization
3.5. Rad52-V95I Affects the N-terminal ssDNA Binding Domain, But Does Not Impair Rad52 Global DNA Binding and Only Marginally Reduces Pairing of RPA-Coated ssDNA
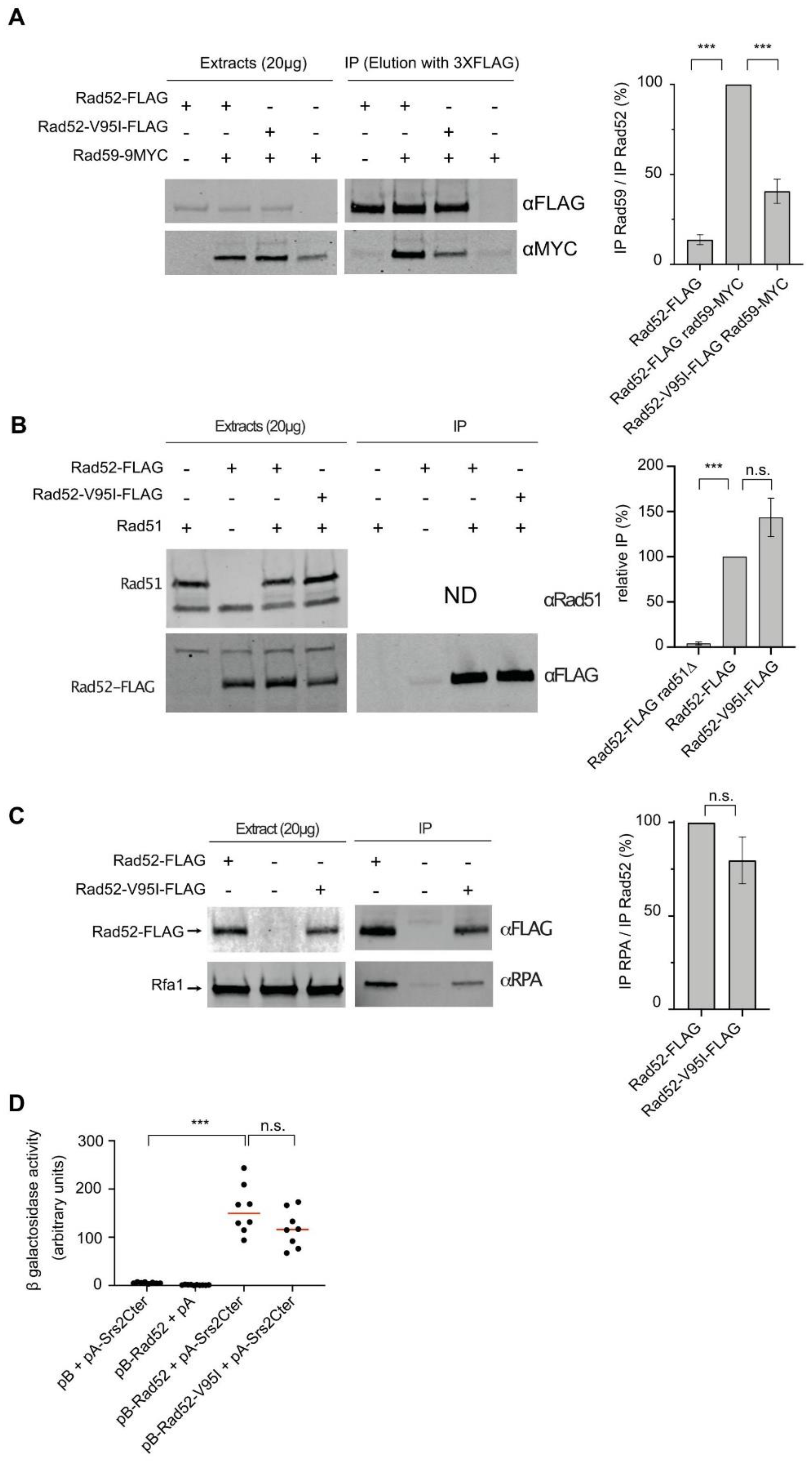
3.6. The V95I Mutation Reduces Rad52 Interaction with Rad59 But Does Not Impair Interaction with RPA, Rad51 and Srs2
3.7. The V95I Mutation Does Not Affect Rad51 Filament Formation at a HO-Induced DSB But Increases Rad51 Filament Disruption by Srs2
3.8. The V95I Mutation Abrogates RPA-Mediated Inhibition of Rad51 Filament Formation In Vitro
3.9. Rad52-V95I Does Not Destabilize Rad51 Filaments in a Competition Assay
4. Discussion
4.1. Rad52 N-Terminal Domain Integrity Is Required for Rad51 Filament Stability
4.2. Rad52-Dependent Stabilization of Rad51 Filaments Can Be Separated from ssDNA Binding and Homologous ssDNA Pairing Activities
4.3. How Does Rad52 Stabilize Rad51 Filaments?
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N. Meiotic Recombination: The Essence of Heredity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016618. [Google Scholar] [CrossRef]
- Zhang, J.-M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Haber, J.E. DNA Repair: The Search for Homology. BioEssays 2018, 40, e1700229. [Google Scholar] [CrossRef] [PubMed]
- Guirouilh-Barbat, J.; Lambert, S.; Bertrand, P.; Lopez, B.S. Is homologous recombination really an error-free process? Front. Genet. 2014, 5, 175. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Rothstein, R.; Lisby, M. Mechanisms and Regulation of Mitotic Recombination in Saccharomyces cerevisiae. Genetics 2014, 198, 795–835. [Google Scholar] [CrossRef] [PubMed]
- Spies, M.; Fishel, R. Mismatch Repair during Homologous and Homeologous Recombination. Cold Spring Harb. Perspect. Biol. 2015, 7, a022657. [Google Scholar] [CrossRef]
- Dupaigne, P.; Le Breton, C.; Fabre, F.; Gangloff, S.; Le Cam, E.; Veaute, X. The Srs2 Helicase Activity Is Stimulated by Rad51 Filaments on dsDNA: Implications for Crossover Incidence during Mitotic Recombination. Mol. Cell 2008, 29, 243–254. [Google Scholar] [CrossRef]
- Ira, G.; Malkova, A.; Liberi, G.; Foiani, M.; Haber, J.E. Srs2 and Sgs1–Top3 Suppress Crossovers during Double-Strand Break Repair in Yeast. Cell 2003, 115, 401–411. [Google Scholar] [CrossRef]
- Mitchel, K.; Lehner, K.; Jinks-Robertson, S. Heteroduplex DNA Position Defines the Roles of the Sgs1, Srs2, and Mph1 Helicases in Promoting Distinct Recombination Outcomes. PLoS Genet. 2013, 9, e1003340. [Google Scholar] [CrossRef] [PubMed]
- Mazón, G.; Symington, L.S. Mph1 and Mus81-Mms4 Prevent Aberrant Processing of Mitotic Recombination Intermediates. Mol. Cell 2013, 52, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Satory, D.; Dray, E.; Papusha, A.; Scheller, J.; Kramer, W.; Krejci, L.; Klein, H.; Haber, J.E.; Sung, P.; et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: Implications for crossover control in mitotic recombination. Genes Dev. 2009, 23, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P.; Plank, J.L.; Bachrati, C.Z.; Hickson, I.D.; Kowalczykowski, S.C. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1–Top3. Nat. Struct. Mol. Biol. 2010, 17, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Hickson, I.D. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nat. Cell Biol. 2003, 426, 870–874. [Google Scholar] [CrossRef]
- Carver, A.; Zhang, X. Rad51 filament dynamics and its antagonistic modulators. Semin. Cell Dev. Biol. 2021, 113, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Belan, O.; Barroso, C.; Kaczmarczyk, A.; Anand, R.; Federico, S.; O’Reilly, N.; Newton, M.D.; Maeots, E.; Enchev, R.I.; Martinez-Perez, E.; et al. Single-molecule analysis reveals cooperative stimulation of Rad51 filament nucleation and growth by mediator proteins. Mol. Cell 2021, 81, 1058–1073.e7. [Google Scholar] [CrossRef]
- Candelli, A.; Holthausen, J.T.; Depken, M.; Brouwer, I.; Franker, M.A.M.; Marchetti, M.; Heller, I.; Bernard, S.; Garcin, E.B.; Modesti, M.; et al. Visualization and quantification of nascent RAD51 filament formation at single-monomer resolution. Proc. Natl. Acad. Sci. USA 2014, 111, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Miné, J.; Disseau, L.; Takahashi, M.; Cappello, G.; Dutreix, M.; Viovy, J.-L. Real-time measurements of the nucleation, growth and dissociation of single Rad51–DNA nucleoprotein filaments. Nucleic Acids Res. 2007, 35, 7171–7187. [Google Scholar] [CrossRef]
- Qiu, Y.; Antony, E.; Doganay, S.; Koh, H.R.; Lohman, T.M.; Myong, S. Srs2 prevents Rad51 filament formation by repetitive motion on DNA. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. RAD51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46. [Google Scholar] [CrossRef]
- Bernstein, K.; Reid, R.J.; Sunjevaric, I.; DeMuth, K.; Burgess, R.C.; Rothstein, R. The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti-recombinase. Mol. Biol. Cell 2011, 22, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.C.; Lisby, M.; Altmannova, V.; Krejci, L.; Sung, P.; Rothstein, R. Localization of recombination proteins and Srs2 reveals anti-recombinase function in vivo. J. Cell Biol. 2009, 185, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nat. Cell Biol. 2003, 423, 305–309. [Google Scholar] [CrossRef]
- Liu, J.; Renault, L.; Veaute, X.; Fabre, F.; Stahlberg, H.; Heyer, W.D. Rad51 paralogues Rad55-Rad57 balance the antirecombinase Srs2 in Rad51 filament formation. Nature 2011, 479, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; Dupaigne, P.; Maloisel, L.; Guerois, R.; Le Cam, E.; Coïc, E. Rad52-Rad51 association is essential to protect Rad51 filaments against Srs2, but facultative for filament formation. eLife 2018, 7, e32744. [Google Scholar] [CrossRef] [PubMed]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nat. Cell Biol. 2003, 423, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Esta, A.; Ma, E.; Dupaigne, P.; Maloisel, L.; Guerois, R.; Le Cam, E.; Veaute, X.; Coïc, E. Rad52 Sumoylation Prevents the Toxicity of Unproductive Rad51 Filaments Independently of the Anti-Recombinase Srs2. PLoS Genet. 2013, 9, e1003833. [Google Scholar] [CrossRef] [PubMed]
- Vasianovich, Y.; Altmannova, V.; Kotenko, O.; Newton, M.D.; Krejci, L.; Makovets, S. Unloading of homologous recombination factors is required for restoring double-stranded DNA at damage repair loci. EMBO J. 2017, 36, 213–231. [Google Scholar] [CrossRef]
- Bergink, S.; Ammon, T.; Kern, M.; Schermelleh, L.; Leonhardt, H.; Jentsch, S. Role of Cdc48/p97 as a SUMO-targeted segregase curbing Rad51–Rad52 interaction. Nat. Cell Biol. 2013, 15, 526–532. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Bendixen, C.; Sunjevaric, I.; Rothstein, R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA 1996, 93, 10729–10734. [Google Scholar] [CrossRef] [PubMed]
- Seong, C.; Sehorn, M.G.; Plate, I.; Shi, I.; Song, B.; Chi, P.; Mortensen, U.; Sung, P.; Krejci, L. Molecular anatomy of the recombination mediator function of Saccharomyces cerevisiae Rad52. J. Biol. Chem. 2008, 283, 12166–12174. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, W.; Kurumizaka, H.; Ishitani, R.; Fukai, S.; Nureki, O.; Shibata, T.; Yokoyama, S. Crystal Structure of the Homologous-Pairing Domain from the Human RAD52 Recombinase in the Undecameric Form. Mol. Cell 2002, 10, 359–371. [Google Scholar] [CrossRef]
- Ranatunga, W.; Jackson, D.; Lloyd, J.A.; Forget, A.L.; Knight, K.L.; Borgstahl, G.E.O. Human RAD52 Exhibits Two Modes of Self-association. J. Biol. Chem. 2001, 276, 15876–15880. [Google Scholar] [CrossRef]
- Shinohara, A.; Shinohara, M.; Ohta, T.; Matsuda, S.; Ogawa, T. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 1998, 3, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.R.; Wentzell, L.M.; Liu, Y.; West, S.; Wigley, D.B. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. USA 2002, 99, 13492–13497. [Google Scholar] [CrossRef]
- Stasiak, A.Z.; Larquet, E.; Stasiak, A.; Müller, S.; Engel, A.; Van Dyck, E.; West, S.; Egelman, E.H. The human RAD52 protein exists as a heptameric ring. Curr. Biol. 2000, 10, 337–340. [Google Scholar] [CrossRef]
- De Mayolo, A.A.; Lisby, M.; Erdeniz, N.; Thybo, T.; Mortensen, U.H.; Rothstein, R. Multiple start codons and phosphorylation result in discrete Rad52 protein species. Nucleic Acids Res. 2006, 34, 2587–2597. [Google Scholar] [CrossRef]
- Hansson, M.D.; Rzeznicka, K.; Rosenbäck, M.; Hansson, M.; Sirijovski, N. PCR-mediated deletion of plasmid DNA. Anal. Biochem. 2008, 375, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Schaffer, A.A.; Aravind, L.; Madden, T.L.; Shavirin, S.; Spouge, J.L.; Wolf, Y.I.; Koonin, E.V.; Altschul, S.F. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res 2001, 29, 2994–3005. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.; Martin, D.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Pupko, T.; Bell, R.E.; Mayrose, I.; Glaser, F.; Ben-Tal, N. Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 2002, 18, S71–S77. [Google Scholar] [CrossRef]
- Strahl-Bolsinger, S.; Hecht, A.; Luo, K.; Grunstein, M. SIR2 and SIR4 interactions differ in core and extended telomeric heterochromatin in yeast. Genes Dev. 1997, 11, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Elledge, S.J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 2007, 4, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kantake, N.; Sugiyama, T.; Kowalczykowski, S.C. Rad51 Protein Controls Rad52-mediated DNA Annealing. J. Biol. Chem. 2008, 283, 14883–14892. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, U.H.; Erdeniz, N.; Feng, Q.; Rothstein, R. A Molecular Genetic Dissection of the Evolutionarily Conserved N Terminus of Yeast Rad52. Genetics 2002, 161, 549–562. [Google Scholar] [CrossRef]
- Shi, I.; Hallwyl, S.C.L.; Seong, C.; Mortensen, U.; Rothstein, R.; Sung, P. Role of the Rad52 Amino-terminal DNA Binding Activity in DNA Strand Capture in Homologous Recombination. J. Biol. Chem. 2009, 284, 33275–33284. [Google Scholar] [CrossRef]
- Heude, M.; Fabre, F. a/alpha-control of DNA repair in the yeast Saccharomyces cerevisiae: Genetic and physiological aspects. Genetics 1993, 133, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Lovett, S.T.; Mortimer, R.K. Characterization of Null Mutants of the RAD55 Gene of Saccharomyces cerevisiae: Effects of Temperature, Osmotic Strength and Mating Type. Genetics 1987, 116, 547–553. [Google Scholar] [CrossRef]
- Mozlin, A.M.; Fung, C.W.; Symington, L.S. Role of the Saccharomyces cerevisiae Rad51 paralogs in sister chromatid recombination. Genetics 2008, 178, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Vaze, M.B.; Pellicioli, A.; Lee, S.E.; Ira, G.; Liberi, G.; Arbel-Eden, A.; Foiani, M.; Haber, J.E. Recovery from Checkpoint-Mediated Arrest after Repair of a Double-Strand Break Requires Srs2 Helicase. Mol. Cell 2002, 10, 373–385. [Google Scholar] [CrossRef]
- Elango, R.; Sheng, Z.; Jackson, J.; Decata, J.; Ibrahim, Y.; Pham, N.T.; Liang, D.H.; Sakofsky, C.J.; Vindigni, A.; Lobachev, K.S.; et al. Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saotome, M.; Saito, K.; Yasuda, T.; Ohtomo, H.; Sugiyama, S.; Nishimura, Y.; Kurumizaka, H.; Kagawa, W. Structural Basis of Homology-Directed DNA Repair Mediated by RAD52. iScience 2018, 3, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Ventura, S.; Vega, M.C.; Lacroix, E.; Angrand, I.; Spagnolo, L.; Serrano, L. Conformational strain in the hydrophobic core and its implications for protein folding and design. Nat. Genet. 2002, 9, 485–493. [Google Scholar] [CrossRef]
- Davis, A.P.; Symington, L.S. The Yeast Recombinational Repair Protein Rad59 Interacts With Rad52 and Stimulates Single-Strand Annealing. Genetics 2001, 159, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sugiyama, T.; Kowalczykowski, S.C. DNA annealing mediated by Rad52 and Rad59 proteins. J. Biol. Chem. 2006, 281, 15441–15449. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, N.; Ira, G.; Haber, J.E. DNA Length Dependence of the Single-Strand Annealing Pathway and the Role of Saccharomyces cerevisiae RAD59 in Double-Strand Break Repair. Mol. Cell. Biol. 2000, 20, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Symington, L.S. A Rad52 homolog is required for Rad51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996, 10, 2025–2037. [Google Scholar] [CrossRef] [PubMed]
- Kolesar, P.; Altmannova, V.; Silva, S.; Lisby, M.; Krejci, L. Pro-recombination role of Srs2 requires SUMO but is independent of PCNA interaction. J. Biol. Chem. 2016, 291, 7594–7607. [Google Scholar] [CrossRef] [PubMed]
- Filippo, J.S.; Sung, P.; Klein, H. Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Broomfield, S.; Xiao, W. Suppression of genetic defects within the RAD6 pathway by srs2 is specific for error-free post-replication repair but not for damage-induced mutagenesis. Nucleic Acids Res. 2002, 30, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Pfander, B.; Moldovan, G.L.; Sacher, M.; Hoege, C.; Jentsch, S. SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 2005, 436, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Gaines, W.A.; Godin, S.K.; Kabbinavar, F.F.; Rao, T.; VanDeMark, A.P.; Sung, P.; Bernstein, K.A. Promotion of presynaptic filament assembly by the ensemble of S. cerevisiae Rad51 paralogues with Rad52. Nat. Commun. 2015, 6, 7834. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, E.; Maloisel, L.; Le Falher, L.; Guérois, R.; Coïc, E. Rad52 Oligomeric N-Terminal Domain Stabilizes Rad51 Nucleoprotein Filaments and Contributes to Their Protection against Srs2. Cells 2021, 10, 1467. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061467
Ma E, Maloisel L, Le Falher L, Guérois R, Coïc E. Rad52 Oligomeric N-Terminal Domain Stabilizes Rad51 Nucleoprotein Filaments and Contributes to Their Protection against Srs2. Cells. 2021; 10(6):1467. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061467
Chicago/Turabian StyleMa, Emilie, Laurent Maloisel, Léa Le Falher, Raphaël Guérois, and Eric Coïc. 2021. "Rad52 Oligomeric N-Terminal Domain Stabilizes Rad51 Nucleoprotein Filaments and Contributes to Their Protection against Srs2" Cells 10, no. 6: 1467. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061467