
Fabry Cardiomyopathy: Current Practice and Future Directions


1
Department of Medicine, University of British Columbia, Vancouver, BC V6H 0A5, Canada
2
Faculty of Medicine, University of British Columbia, Vancouver, BC V6H 0A5, Canada
3
Vancouver General Hospital and University of British Columbia Echocardiography Laboratory, Division of Cardiology, University of British Columbia, Vancouver, BC V6H 0A5, Canada
*
Authors to whom correspondence should be addressed.
†
These authors are co-senior authors.
Cells 2021, 10(6), 1532; https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061532
Submission received: 6 May 2021
/
Revised: 15 June 2021
/
Accepted: 15 June 2021
/
Published: 17 June 2021
(This article belongs to the Special Issue Lysosomal Storage Disorders)
Abstract
:Fabry disease (FD) is an X-linked lysosomal storage disorder caused by mutations in the galactosidase A (GLA) gene that result in deficient galactosidase A enzyme and subsequent accumulation of glycosphingolipids throughout the body. The result is a multi-system disorder characterized by cutaneous, corneal, cardiac, renal, and neurological manifestations. Increased left ventricular wall thickness represents the predominant cardiac manifestation of FD. As the disease progresses, patients may develop arrhythmias, advanced conduction abnormalities, and heart failure. Cardiac biomarkers, point-of-care dried blood spot testing, and advanced imaging modalities including echocardiography with strain imaging and magnetic resonance imaging (MRI) with T1 mapping now allow us to detect Fabry cardiomyopathy much more effectively than in the past. While enzyme replacement therapy (ERT) has been the mainstay of treatment, several promising therapies are now in development, making early diagnosis of FD even more crucial. Ongoing initiatives involving artificial intelligence (AI)-empowered interpretation of echocardiographic images, point-of-care dried blood spot testing in the echocardiography laboratory, and widespread dissemination of point-of-care ultrasound devices to community practices to promote screening may lead to more timely diagnosis of FD. Fabry disease should no longer be considered a rare, untreatable disease, but one that can be effectively identified and treated at an early stage before the development of irreversible end-organ damage.
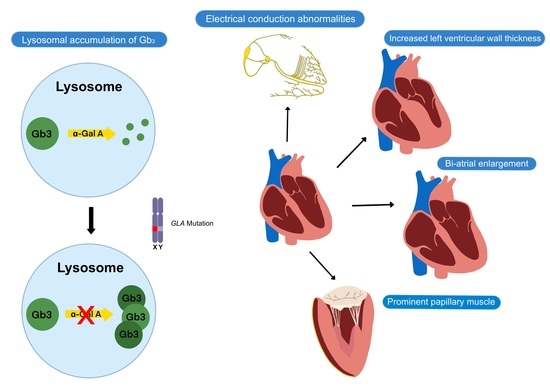
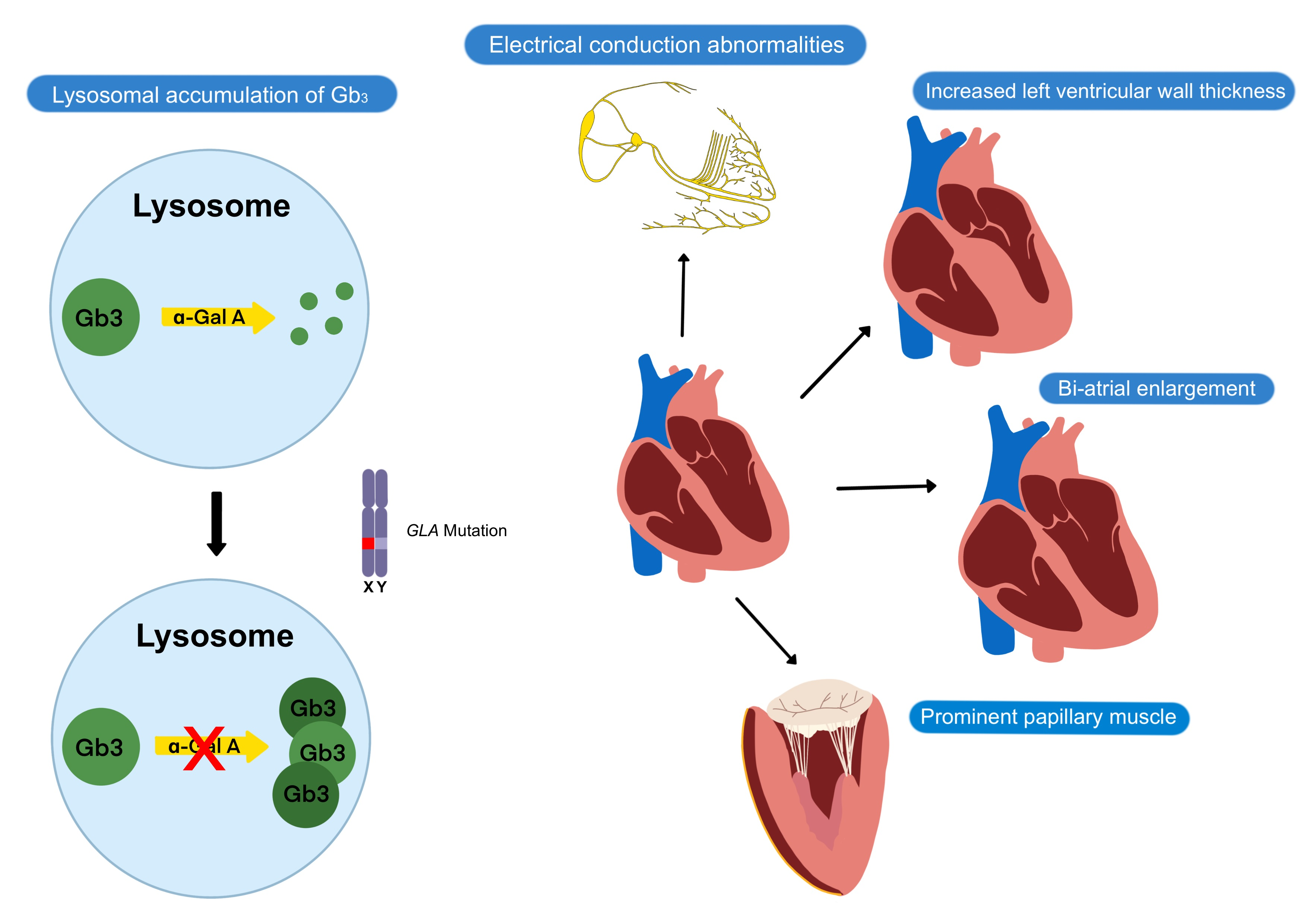
1. Introduction
Fabry disease is an X-linked lysosomal storage disorder caused by mutations in the α-galactosidase A (GLA) gene that result in deficient α-galactosidase A (α-Gal A) enzyme and the accumulation of globotriaosylceramide (Gb3) and associated glycosphingolipids throughout the body [1]. The accumulation of Gb3 in lysosomes leads to metabolic dysfunction and subsequent cellular death in various organs, leading to a multisystemic clinical presentation that includes cutaneous, corneal, renal, neurological, and cardiac manifestations.
Fabry disease has long been considered a rare disease with limited diagnostic and treatment options. However, the condition is likely underdiagnosed, with newborn screening programs around the world showing much higher prevalence of GLA mutation than previously described [2,3]. Furthermore, we now have more effective tools to diagnose FD in a more timely fashion [4,5,6,7]. The development of novel effective therapies has made the early diagnosis of FD and prompt institution of therapy even more important.
The purpose of this review is to provide an overview of the cardiac manifestations of Fabry disease, methods of screening and diagnosis, currently available and investigational treatments, ongoing challenges in management, and future directions to improve the care of patients with this condition.
2. Clinical Presentation of Fabry Disease
The typical presentation of Type I ‘classical FD’ is a male patient of the first and second decades of life who presents with acroparesthesia (burning pain in the extremities), gastrointestinal symptoms (including nausea, diarrhea or constipation, and abdominal pain), and angiokeratoma corporis (distinct cutaneous abnormality characterized by vascular papules distributed in the inguinal, hip, and periumbilical areas) [8]. Patients subsequently develop more severe cardiac, renal, and neurologic complications in the third and fourth decades of life [8]. Classical FD has a typical disease onset of childhood or early adolescence and is described in hemizygous male FD patients or heterozygous female FD patients with skewed X-chromosome inactivation of the normal GLA allele [9]. Classical FD patients are characterized by a nearly absent level of α-Gal A activity. In contrast to other lysosomal storage diseases, a large number of patients with FD have the late-onset Type II ‘non-classical FD’ phenotype, remaining asymptomatic during very first few decades of life due to residual α-Gal A activity. Recently, sub-classifications of FD including “cardiac variants” with isolated cardiac findings have been identified [10,11].
3. Fabry Cardiomyopathy
Cardiac manifestations of FD include increased left ventricular (LV) wall thickness, conduction abnormalities, arrhythmias, valvular disease, and aortic dilatation, which result from glycolipid deposition and subsequent fibrosis of contractile cardiomyocytes, conductive cardiomyocytes, valvular interstitial cells, and smooth muscle cells of the cardiovascular system (Table 1). Eventually, complications such as hypertension, myocardial infarction, and cardiac death may occur, with heart failure being the most common first cardiovascular event in FD [12]. Compared to other organs, the heart appears to be the most susceptible to low levels of α-Gal A. The FD-related cardiovascular injury is thought to be due to a combination of Gb3 accumulation, the accumulation of trophic factors, and microcirculatory ischemia, which contribute to inflammation and ultimately result in myocardial fibrosis [13]. Patients with FD-related cardiac involvement tend to be asymptomatic from a cardiac perspective during the first four decades of life, then present with non-specific cardiac symptoms such as angina, dyspnea, palpitations, or syncope. Since there is no pathognomonic cardiac manifestation of FD, the non-specific findings often make FD-related cardiac involvement difficult to diagnose.
The hallmark feature of FD cardiomyopathy is increased LV wall thickness [6,9]. Increased right ventricular wall thickness and impaired right ventricular function have also been reported [16]. Increased LV wall thickness is rarely present in children with FD, tends to be more severe in male FD patients, and is usually not evident until the third or fourth decade in classical FD patients [4,17]. However, the finding of increased LV wall thickness is not specific for Fabry cardiomyopathy, and it is important for clinicians to consider the differential diagnoses of other causes of increased LV wall thickness (Table 2).
Electrophysiologic abnormalities represent other common cardiac manifestations of FD [9]. Advanced conduction disease is thought to be caused by glycolipid accumulation in cardiomyocytes of the atrioventricular (AV) node, bundle of His, and the left and right bundle branches [18]. In contrast, accelerated AV conduction is common in younger FD patients and is reflected as shortened PR intervals on the electrocardiogram (ECG), while prolonged PR interval may be observed in older FD patients [18]. Atrial and ventricular arrhythmias are also relatively common and may be due to atrial myopathy, atrial dilatation from longstanding diastolic dysfunction, and atrial and ventricular fibrosis. Atrial arrhythmias such as atrial fibrillation are more common than ventricular arrhythmias.
In addition, valvular diseases such as aortic, mitral, and tricuspid regurgitation are common in patients with FD due to mild thickening of the valves, although valvular regurgitation significant enough to require intervention is uncommon and stenotic lesions attributed to Fabry disease alone are rare [19]. Thickening of papillary muscles in FD patients has been proposed as a mechanism of mitral regurgitation in FD. Fabry disease can also lead to aortic dilatation, especially in males, where its prevalence increases with age. Aortic dilatation in FD has been shown to be independent of elevated blood pressure [20] and has been attributed to degenerative changes in the aortic media due to excessive glycolipid substrate deposition [21]. Significant aortic dilatation due to Fabry disease resulting in acute aortic events has yet to be reported.
Fabry cardiomyopathy may not be as rare as we once thought as it has been shown to be responsible for up to 4% of unexplained hypertrophic cardiomyopathy (HCM) cases [22,23] and up to 12% of unexplained increased LV wall thickness in other selected cohorts [9]. When assessing for pathogenic mutations only, the prevalence of GLA mutation in LVH or HCM clinics is 0.94% in males and 0.90% in females [24]. In fact, the cardiac variant is the most common form of FD in some countries such as Taiwan [25]. This is of particular concern as cardiovascular complications represent the predominant source of FD-related mortality and morbidity [8,26].
4. Screening and Diagnosis
A diagnosis of FD is made by demonstrating reduced or absent α-Gal A activity in hemizygous males. In females, genotyping is required as random inactivation of X-chromosome results in mosaicism, resulting in partial expression of the mutated allele that allows for normal levels of α-Gal A activity but still results in Gb3 build-up [27].
The prevalence of FD in males was previously estimated to be 1 in 117,000 [28]. However, various newborn screening initiatives around the world such as in Taiwan and Italy have demonstrated a much higher prevalence of disease-causing variants, ranging from 1:1250 to 1:4600, suggesting that FD may be underdiagnosed elsewhere [2,3].
The screening and diagnosis of FD have been simplified with the use of dried blood spot (DBS) testing. Dried blood testing identifies reduced enzyme activity using artificial fluorescent tag substrates linked to an analog of the natural substrate [7]. If enzyme activity is found to be low in male patients, a confirmatory genetic analysis is sent. For female patients, enzyme activity is not a reliable measure of disease activity and therefore all DBS samples are sent for genetic analysis.
5. Diagnosis of Fabry Cardiomyopathy
Awareness of the cardiac manifestations of FD may lead to earlier recognition of the condition and differentiation from other causes of increased LV wall thickness. Sensitive cardiac biomarkers and advanced cardiac imaging modalities such as echocardiography with strain imaging and MRI with T1 mapping are essential for the diagnosis and staging of FD.
5.1. Echocardiography
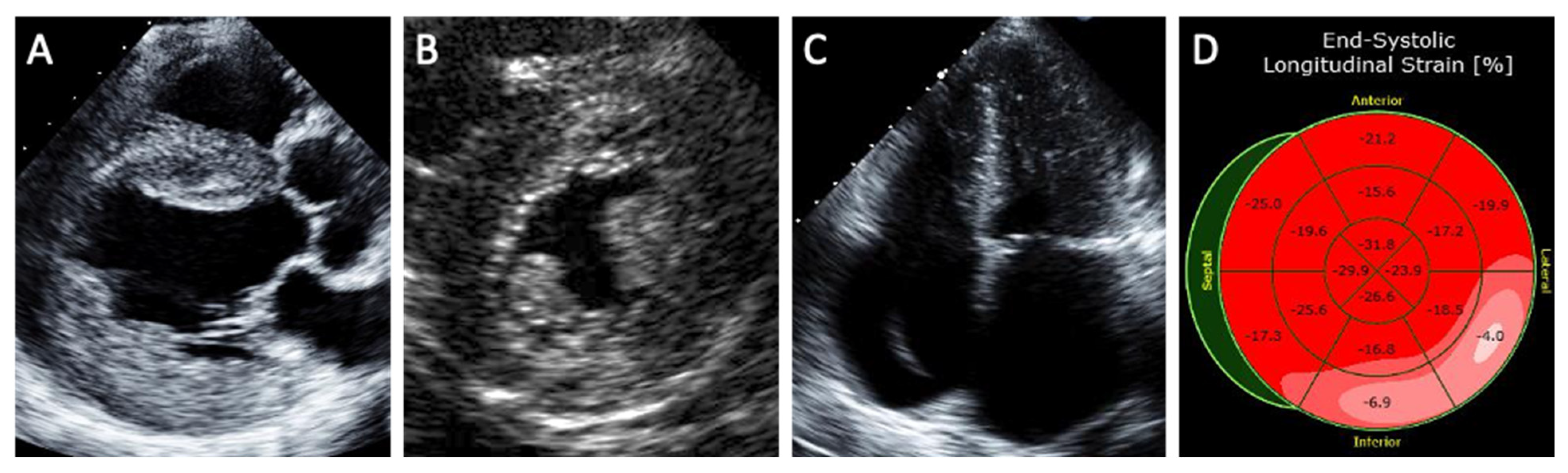
Echocardiography is an effective noninvasive method of assessing the degree of cardiac involvement in FD (Figure 1). A concentric pattern of increased LV wall thickness is the hallmark finding of Fabry cardiomyopathy, although other morphologies such as an asymmetric thickening of the interventricular septum, eccentric hypertrophy, and apical hypertrophy have also been described [4]. Other echocardiographic features of Fabry cardiomyopathy include prominent papillary muscles, increased right ventricular wall thickness, atrial enlargement, and the ‘binary sign’. The ‘binary sign’ is a finding characterized by a hyperechogenic endocardial surface composed of glycolipid-enriched smooth muscle cells adjacent to a hypoechogenic subendocardial layer relatively devoid of glycolipids, although recent studies have shown poor sensitivity and specificity of the sign to detect FD [29].
Strain imaging is a sensitive method in identifying subclinical cardiomyopathy. Patients with Fabry cardiomyopathy demonstrate lower global longitudinal strain and circumferential strain compared to healthy subjects [30]. Reduced longitudinal strain in the basal inferolateral segment as well as loss of the base-to-apex circumferential strain gradient have been suggested as specific LV deformation patterns of Fabry cardiomyopathy compared to hypertrophic cardiomyopathy [31].
The application of artificial intelligence (AI) to the echo assessment of patients with increased LV wall thickness may one day facilitate the diagnosis of FD given the known challenges of accurate LV wall thickness measurement [32]. Artificial intelligence-based myocardial texture analysis was suggested as a means of differentiating hypertrophic cardiomyopathy from hypertensive heart disease and uremic cardiomyopathy [33]. Artificial intelligence models have also previously been shown to augment the detection of cardiac amyloidosis and assist in the diagnosis and risk stratification of patients with hypertrophic cardiomyopathy [34,35,36]. The role of AI in assessment of patients with increased wall thickness including patients with possible FD is ongoing in our echocardiography laboratory.
5.2. Magnetic Resonance Imaging
Several MRI findings have been described in Fabry cardiomyopathy. Late gadolinium enhancement (LGE) in the basal inferolateral segment is a common MRI finding in Fabry cardiomyopathy and is observed in 50% of affected patients [5]. Shortened myocardial T1 relaxation time can discriminate Fabry cardiomyopathy from other causes of LVH and may be seen in Fabry cardiomyopathy prior to the development of LVH [37,38]. Chronic local T2 elevation in the basal inferolateral segment may indicate myocardial inflammation from Fabry cardiomyopathy and is associated with worse Fabry stabilization index (FASTEX) score [39]. Cardiac MRI can also be helpful in identifying increased right ventricular wall thickness, atrial enlargement, and prominent papillary muscles.
5.3. Laboratory Tests
Various laboratory biomarkers have been proposed for use in staging patients with Fabry cardiomyopathy. Troponin level has been correlated with the degree of fibrosis measured by LGE on MRI in patients with Fabry Cardiomyopathy [40]. Increased symptom and disease burden is correlated with elevated levels of CRP, NT-proBNP, and IL-6 [41,42,43].
5.4. Cardiopulmonary Exercise Test
Patients with FD have been shown to have decreased heart rate, indexed oxygen pulse, blood pressure, and max VO2 at peak exercise during cardiopulmonary exercise testing using treadmill test and cycle ergometer [44,45,46]. There may be a small improvement in exercise tolerance in patients receiving ERT [44,47].
6. Fabry Disease Severity Scores
Several validated scoring systems for FD is available. Fabry disease severity scoring system (DS3) is a validated scoring system utilizing 4 clinical domains: peripheral nervous system, renal, cardiac, and a patient-reported domain. Fabry DS3 has been demonstrated to correlate very well with overall clinical picture of patients with FD using clinical global impression of severity score by FD experts [48]. The Mainz Severity Score Index (MSSI) is another scoring system used to grade severity of disease in FD [49]. The MSSI is composed of four sections (general, neurological, cardiac, and renal) related to symptoms of FD. After one year of treatment with ERT in patients with FD, MSSI was significantly reduced in all patients [49].
The Fabry Stabilization index (FASTEX) is a scoring system developed to assess for clinical stability in patients with FD. The FASTEX was created using consensus weighted score in 28 patients with FD, where the score is based on three domains (nervous system, renal, and cardiac). A worsening global score of ≥20% was suggested to indicate that the patient is clinically unstable [50].
7. Treatments in Fabry Disease
The current approach to the treatment and management of FD aims to either prevent or delay the progression of FD to irreversible tissue damage and organ failure. There is currently no curative treatment for FD. To date, treatments available for FD include disease-modifying therapies used in conjunction with non-specific therapies that treat symptoms caused by multi-organ injury. The advantages and disadvantages of currently available as well as investigational FD therapies are summarized in Table 3.
Enzyme replacement therapy (ERT) is currently the standard treatment for males with classical FD and Type 2 non-classical FD, and females with classical FD. Enzyme replacement therapy became available in 2001 and represents the first treatment developed for FD. Two formulations of ERT currently exist: agalsidase α (Replagal) administered at a dose of 0.2 mg/kg intravenously every two weeks and agalsidase β (Fabrazyme) administered at a dose of 1 mg/kg intravenously every two weeks. Agalsidase α is generated from a continuous human cell line with the activation of the GLA gene, while agalsidase β is generated from a Chinese hamster ovary mammalian cell expression system transduced with the human GLA sequence. Enzyme replacement therapy has been shown to effectively reduce glycolipid substrates including Gb3 in the urine, plasma, and tissues of patients with FD [51]. With respect to FD-related cardiac injury, ERT has been shown to effectively reduce Gb3 inclusions in endothelial cells, with less clear evidence regarding Gb3 clearance from cardiomyocytes [52]. In addition, observational studies have reported a reduction in LV wall thickness in patients treated with ERT [53,54].
The limitations of ERT include the short plasma half-life of the recombinant enzyme, thus necessitating bi-weekly infusions and that it can only delay the progression of FD. Enzyme replacement therapy also has limited efficacy in later stages of Fabry cardiomyopathy, when fibrosis is already present [53], and it is unclear whether or not ERT slows the progression of fibrosis [55]. Additionally, anti-drug antibodies against the recombinant replacement enzyme in ERT has been reported to develop in 64–88% of FD patients [52,56], thereby attenuating the effect of ERT. Finally, ERT demonstrates uneven biodistribution, with the liver taking up the majority of the recombinant replacement enzyme, whereas the most severely affected cell types in the body such as cardiomyocytes and podocytes take up lesser amounts of the replacement enzyme [55].
There is currently no evidence demonstrating the superiority of agalsidase α over agalsidase β and vice versa in clinical endpoints [57,58]. Specifically, in the Canadian Fabry Disease Initiative, a comparison of agalsidase α and agalsidase β demonstrated no statistical difference in clinical endpoints including death, cardiac events, acute neurological events, and others [58]. However, there were differences in the biochemical response between patients treated with agalsidase α and agalsidase β, with a higher risk of developing anti-drug antibodies and a greater decrease in the plasma globotriaosylsphingosine levels in patients treated with agalsidase β. In addition, there was a greater reduction in the left ventricular mass in patients treated with agalsidase β [59].
Oral pharmacologic chaperone therapy, namely Migalastat, is an alternative treatment option for FD. However, since Migalastat is protein-variant specific, it is only used for patients with amenable GLA gene variants [60]. These specific GLA variants produce highly unstable mutated α-Gal A proteins. Migalastat binds to these α-Gal A variants, thus stabilizing the enzymes by enhancing correct folding [55]. This stabilization allows the mutated enzymes to be properly trafficked to lysosomes, where Migalastat dissociates, allowing it to catabolize the accumulated Gb3 substrates [61]. This therapy has been demonstrated to both increase α-Gal A activity and decrease Gb3 inclusions [62,63].
Despite disease-modifying FD treatments described, equal attention and care should be given to non-FD specific treatments directed towards the multi-system consequences of the condition. Due to the clinical heterogeneity of FD, a multidisciplinary clinical team with a cardiologist, nephrologist, neurologist, genetic counselor, and a medical geneticist should ideally be in place for the holistic care of FD patients. General preventative measures including pharmacological stroke prophylaxis with an antithrombic agent and lifestyle modifications such as avoidance of extremes of temperature to prevent painful crises, exercise prescription, diet, and smoking cessation should be appropriately advised. Other co-morbidities such as hypertension and dyslipidemia should be managed diligently. The management of the cardiac manifestations of FD has been summarized in Table 4.
8. Future Directions in the Management of Fabry Disease
There are three potential future treatment options currently undergoing preclinical investigation: (1) second-generation ERT; (2) substrate reduction therapy (SRT); and (3) gene therapy.
To date, two formulations of second-generation ERTs have been developed: pegunigalsidase-α and moss-aGal. These second-generation ERTs are plant-derived with different pharmacokinetic properties that may lead to better biodistribution in the body compared with first-generation ERTs.
Substrate reduction therapy including Venglustat and Lucerastat are potential future oral treatments for FD. Substrate reduction therapy limits the formation of pathogenic metabolites such as Gb3, thus limiting its accumulation in tissues throughout the body [64]. Substrate reduction therapy may have a potential role as an adjunctive therapy used alongside ERT [64].
Gene therapy may be a treatment option that enables FD patients to receive fewer treatments with more permanent effects. This novel therapy involves the encapsulation of mRNA into lipid nanoparticles that target hepatocytes where endogenous protein translation, glycosylation, and intracellular trafficking of α-Gal A occurs. Functional α-Gal A enzymes are then subsequently secreted into the circulation, which can be taken up by affected cells [65]. Recently, the first gene therapy trial for FD was conducted, where classical FD patients were infused with lentivirus-transduced hematopoietic stem cells engineered to express α-Gal A [51]. All patients produced α-Gal A to near normal levels within one week of therapy with observations of reduced plasma and urine Gb3 [51].
Several challenges exist in the early identification of individuals with FD. Many non-classic FD patients are asymptomatic in their early years of life, resulting in delayed diagnosis. Additionally, remote centers may not have the capacity for advanced imaging. Even when presented with imaging findings of FD, clinicians may not readily diagnose FD due to the non-specific findings and unfamiliarity with the clinical manifestations of FD.
To overcome these challenges, our echocardiography lab has undertaken several initiatives that aim to achieve earlier detection of Fabry cardiomyopathy, including the development of artificial intelligence (AI)-empowered detection of increased LV wall thickness, the use of DBS testing at the time of echocardiography when an unexplained increase in LV wall thickness is identified, and the dissemination of point-of-care ultrasound devices to community healthcare providers to promote widespread screening for the presence of cardiomyopathies [66,67,68].
The use of AI to effectively identify increased LV wall thickness and differentiate between the many possible etiologies is a promising area for further research. An AI approach based on 12-lead ECG has been shown to be 87% sensitive and 90% specific in identifying hypertrophic cardiomyopathy [69]. Fully automated echocardiographic interpretation has also been shown to be capable of detecting hypertrophic cardiomyopathy and cardiac amyloidosis with C-statistics of 0.93 and 0.87 respectively [36]. An AI strategy based on late gadolinium enhancement (LGE) patterns on cardiac MRI demonstrated 88% diagnostic accuracy in detecting cardiac amyloidosis [70]. Further research is needed to determine the potential role of AI in the diagnosis of Fabry disease.
9. Conclusions and Call to Action
Our understanding of the pathophysiology, diagnosis and treatment of FD continue to rapidly evolve. We now have not only the capability to more effectively diagnose FD using various laboratory and imaging modalities, but also effective treatment options for the condition. Clinicians should have an understanding of the clinical manifestations of FD and consider it as part of the differential diagnosis when presented with unexplained increased LV wall thickness. Fabry disease should no longer be considered a rare, untreatable disease, but one that can and should be identified and treated in a timely manner.
Author Contributions
Conceptualization, D.F.Y. and T.S.M.T.; writing—original draft preparation, J.Y. and O.Y.; writing—review and editing, D.F.Y. and T.S.M.T.; supervision, D.F.Y. and T.S.M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to thank Jamie Hyerim Chun for creating the illustration for the graphical abstract.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| a-Gal A | α-galactosidase A |
| ACE | Angiotensin converting enzyme |
| ARB | Angiotensin II receptor blocker |
| AI | Artificial intelligence |
| AV | Atrioventricular |
| DBS | Dried blood spot |
| ECG | Electrocardiogram |
| ERT | Enzyme replacement therapy |
| FD | Fabry disease |
| Gb3 | Globotriaosylceramide |
| HCM | Hypertrophic cardiomyopathy |
| ICD | Implantable cardioverter-defibrillator |
| LGE | Late gadolinium enhancement |
| LV | Left ventricular |
| LVEF | Left ventricular ejection fraction |
| LVOT | Left ventricular outflow tract |
| MRI | Magnetic resonance imaging |
| RV | Right ventricular |
| SRT | Substrate reduction therapy |
| VT | Ventricular tachycardia |
References
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for fabry disease in taiwan reveals a high incidence of the later-onset GLA mutation c.936 + 919G > A (IVS4 + 919G > A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Yeung, D.F.; Sirrs, S.; Tsang, M.Y.C.; Gin, K.; Luong, C.; Jue, J.; Nair, P.; Lee, P.K.; Tsang, T.S.M. Echocardiographic Assessment of Patients with Fabry Disease. J. Am. Soc. Echocardiogr. 2018, 31, 639–649.e2. [Google Scholar] [CrossRef]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Seydelmann, N.; Wanner, C.; Störk, S.; Ertl, G.; Weidemann, F. Fabry disease and the heart. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Fabry disease: Enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta 2001, 308, 195–196. [Google Scholar] [CrossRef]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G. Natural course of Fabry disease: Changing pattern of causes of death in FOS-Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef]
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An Atypical Variant of Fabry’s Disease in Men with Left Ventricular Hypertrophy. N. Engl. J. Med. 1995, 333, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.R.; Hung, S.C.; Chang, F.P.; Yu, W.C.; Sung, S.H.; Hsu, C.L.; Dzhagalov, I.; Yang, C.F.; Chu, T.H.; Lee, H.J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence: Experience With a Highly Prevalent Mutation. J. Am. Coll. Cardiol. 2016, 68, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Dabry disease. Am. J. Cardiol. 2005, 96, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Sanchez-Niño, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Omahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Calcagnino, M.; Murphy, E.; Robin, L.; Atul, M.; Derralynn, H.; Perry, M.E. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace 2011, 13, 1781–1788. [Google Scholar] [CrossRef]
- Namdar, M. Electrocardiographic Changes and Arrhythmia in Fabry Disease. Front. Cardiovasc. Med. 2016, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Niemann, M.; Breunig, F.; Beer, M.; Herrmann, S.; Strotmann, J.; Hu, K.; Emmert, A.; Voelker, W.; Ertl, G.; Wanner, C.; et al. The right ventricle in Fabry disease: Natural history and impact of enzyme replacement therapy. Heart 2010, 96, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Linhart, A.; Baehner, F.; Palecek, T.; Wiethoff, C.M.; Miebach, E.; Whybra, C.; Gal, A.; Bultas, J.; Beck, M. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int. J. Cardiol. 2008, 130, 367–373. [Google Scholar] [CrossRef]
- Mehta, J.; Moller, J.H.; Desnick, R.J.; Ph, D. Electrocardiographic and vectorcardiographic abnormalities in Fabry’s disease. Am. Heart J. 1977, 93, 699–705. [Google Scholar] [CrossRef]
- Linhart, A.; Paleček, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupětová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am. Heart J. 2000, 139, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Barbey, F.; Qanadli, S.D.; Juli, C.; Brakch, N.; Palaek, T.; Rizzo, E.; Jeanrenaud, X.; Eckhardt, B.; Linhart, A. Aortic remodelling in Fabry disease. Eur. Heart J. 2010, 31, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnick, R.J.; Blieden, L.C.; Sharp, H.L.; Hofschire, P.J.; Moller, J.H. Cardiac valvular anomalies in Fabry disease. Clinical, morphologic, and biochemical studies. Circulation 1976, 54, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Sachdev, B.; Takenaka, T.; Teraguchi, H.; Tei, C.; Lee, P.; McKenna, W.J.; Elliott, P.M. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002, 105, 1407–1411. [Google Scholar] [CrossRef] [Green Version]
- Maron, M.S.; Xin, W.; Sims, K.B.; Butler, R.; Haas, T.S.; Rowin, E.J.; Desnick, R.J.; Maron, B.J. Identification of Fabry Disease in a Tertiary Referral Cohort of Patients with Hypertrophic Cardiomyopathy. Am. J. Med. 2018, 131, 200.e1–200.e8. [Google Scholar] [CrossRef]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chong, K.W.; Hsu, J.H.; Yu, H.C.; Shih, C.C.; Huang, C.H.; Lin, S.J.; Chen, C.H.; Chiang, C.C.; Ho, H.J.; et al. High incidence of the cardiac variant of fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2009, 2, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in Fabry disease: A systematic review of risk factors in clinical practice. Europace 2018, 20, f153–f161. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. J. Am. Med. Assoc. 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Mundigler, G.; Gaggl, M.; Heinze, G.; Graf, S.; Zehetgruber, M.; Lajic, N.; Voigtlander, T.; Mannhalter, C.; Sunder-Plassmann, R.; Paschke, E.; et al. The endocardial binary appearance (‘binary sign’) is an unreliable marker for echocardiographic detection of Fabry disease in patients with left ventricular hypertrophy. Eur. J. Echocardiogr. 2011, 12, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Gruner, C.; Verocai, F.; Carasso, S.; Vannan, M.A.; Jamorski, M.; Clarke, J.T.R.; Care, M.; Iwanochko, R.M.; Rakowski, H. Systolic myocardial mechanics in patients with Anderson-Fabry disease with and without left ventricular hypertrophy and in comparison to nonobstructive hypertrophic cardiomyopathy. Echocardiography 2012, 29, 810–817. [Google Scholar] [CrossRef]
- Labombarda, F.; Saloux, E.; Milesi, G.; Bienvenu, B. Loss of base-to-apex circumferential strain gradient: A specific pattern of Fabry cardiomyopathy? Echocardiography 2017, 34, 504–510. [Google Scholar] [CrossRef]
- Hindieh, W.; Weissler-Snir, A.; Hammer, H.; Adler, A.; Rakowski, H.; Chan, R.H. Discrepant Measurements of Maximal Left Ventricular Wall Thickness Between Cardiac Magnetic Resonance Imaging and Echocardiography in Patients With Hypertrophic Cardiomyopathy. Circ. Cardiovasc. Imaging 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Huang, H.; Yu, Q.; Ma, Y.; Zhang, Q.; Zhang, B. Artificial intelligence-based myocardial texture analysis in etiological differentiation of left ventricular hypertrophy. Ann. Transl. Med. 2021, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Mahara, K.; Beussink-Nelson, L.; Ikura, H.; Katsumata, Y.; Endo, J.; Gaggin, H.K.; Shah, S.J.; Itabashi, Y.; MacRae, C.A.; et al. Artificial intelligence-enabled fully automated detection of cardiac amyloidosis using electrocardiograms and echocardiograms. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Augusto, J.B.; Davies, R.H.; Bhuva, A.N.; Knott, K.D.; Seraphim, A.; Alfarih, M.; Lau, C.; Hughes, R.K.; Lopes, L.R.; Shiwani, H.; et al. Diagnosis and risk stratification in hypertrophic cardiomyopathy using machine learning wall thickness measurement: A comparison with human test-retest performance. Lancet Digit. Health 2021, 3, e20–e28. [Google Scholar] [CrossRef]
- Zhang, J.; Deo, R.C. Response by Zhang and Deo to Letter Regarding Article, “Fully Automated Echocardiogram Interpretation in Clinical Practice: Feasibility and Diagnostic Accuracy”. Circulation 2019, 139, 1648–1649. [Google Scholar] [CrossRef]
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and assessment of anderson-fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ. Cardiovasc. Imaging 2013, 6, 392–398. [Google Scholar] [CrossRef] [Green Version]
- Pica, S.; Sado, D.M.; Maestrini, V.; Fontana, M.; White, S.K.; Treibel, T.; Captur, G.; Anderson, S.; Piechnik, S.K.; Robson, M.D.; et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 99. [Google Scholar] [CrossRef]
- Augusto, J.B.; Nordin, S.; Vijapurapu, R.; Baig, S.; Bulluck, H.; Castelletti, S.; Alfarih, M.; Knott, K.; Captur, G.; Kotecha, T.; et al. Myocardial edema, myocyte injury, and disease severity in Fabry disease. Circ. Cardiovasc. Imaging 2020, 13, 10171. [Google Scholar] [CrossRef]
- Seydelmann, N.; Liu, D.; Krämer, J.; Drechsler, C.; Hu, K.; Nordbeck, P.; Schneider, A.; Störk, S.; Bijnens, B.; Ertl, G.; et al. High-sensitivity troponin: A clinical blood biomarker for staging cardiomyopathy in fabry disease. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated inflammatory plasma biomarkers in patients with fabry disease: A critical link to heart failure with preserved ejection fraction. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altarescu, G.; Chicco, G.; Whybra, C.; Delgado-Sanchez, S.; Sharon, N.; Beck, M.; Elstein, D. Correlation between interleukin-6 promoter and C-reactive protein (CRP) polymorphisms and CRP levels with the Mainz Severity Score Index for Fabry disease. J. Inherit. Metab. Dis. 2008, 31, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Coats, C.J.; Parisi, V.; Ramos, M.; Janagarajan, K.; O’Mahony, C.; Dawnay, A.; Lachmann, R.H.; Murphy, E.; Mehta, A.; Hughes, D.; et al. Role of serum N-terminal pro-brain natriuretic peptide measurement in diagnosis of cardiac involvement in patients with anderson-fabry disease. Am. J. Cardiol. 2013, 111, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Lobo, T.; Morgan, J.; Bjorksten, A.; Nicholls, K.; Grigg, L.; Centra, E.; Becker, G. Cardiovascular testing in Fabry disease: Exercise capacity reduction, chronotropic incompetence and improved anaerobic threshold after enzyme replacement. Intern. Med. J. 2008, 38, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Bierer, G.; Kamangar, N.; Balte, D.; Wilcox, W.R.; Mosenifar, Z. Cardiopulmonary exercise testing in fabry disease. Respiration 2005, 72, 504–511. [Google Scholar] [CrossRef]
- Powell, A.W.; Jefferies, J.L.; Hopkin, R.J.; Mays, W.A.; Goa, Z.; Chin, C. Cardiopulmonary fitness assessment on maximal and submaximal exercise testing in patients with Fabry disease. Am. J. Med. Genet. Part A 2018, 176, 1852–1857. [Google Scholar] [CrossRef]
- Bierer, G.; Balfe, D.; Wilcox, W.R.; Mosenifar, Z. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J. Inherit. Metab. Dis. 2006, 29, 572–579. [Google Scholar] [CrossRef]
- Giannini, E.H.; Mehta, A.B.; Hilz, M.J.; Beck, M.; Bichet, D.G.; Brady, R.O.; West, M.; Germain, D.P.; Wanner, C.; Waldek, S.; et al. A validated disease severity scoring system for Fabry disease. Mol. Genet. Metab. 2010, 99, 283–290. [Google Scholar] [CrossRef]
- Whybra, C.; Kampmann, C.; Krummenauer, F.; Ries, M.; Mengel, E.; Miebach, E.; Baehner, F.; Kim, K.; Bajbouj, M.; Schwarting, A.; et al. The Mainz Severity Score Index: A new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy. Clin. Genet. 2004, 65, 299–307. [Google Scholar] [CrossRef]
- Mignani, R.; Pieruzzi, F.; Berri, F.; Burlina, A.; Chinea, B.; Gallieni, M.; Pieroni, M.; Salviati, A.; Spada, M. FAbry STabilization indEX (FASTEX): An innovative tool for the assessment of clinical stabilization in Fabry disease. Clin. Kidney J. 2016, 9, 739–747. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Eng, C.M.; Guffon, N.; Wilcox, W.R.; Germain, D.P.; Lee, P.; Waldek, S.; Caplan, L.; Linthorst, G.E.; Desnick, R.J. Safety and Efficacy of Recombinant Human α-Galactosidase A Replacement Therapy in Fabry’s Disease. N. Engl. J. Med. 2001, 345, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Störk, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy. Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Turschner, O.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. Improvement of cardiac function during enzyme replacement therapy in patients with fabry disease: A prospective strain rate imaging study. Circulation 2003, 108, 1299–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Veen, S.J.; Hollak, C.E.M.; Van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, R.; Kopp, J.B.; Austin, H.A.; Balow, J.E.; Brady, R.O. Enzyme Replacement Therapy in Fabry Disease: A Randomized Controlled Trial. N. Engl. J. Med. 2001, 285, 2743. [Google Scholar] [CrossRef] [PubMed]
- El Dib, R.; Gomaa, H.; Carvalho, R.P.; Camargo, S.E.; Bazan, R.; Barretti, P.; Barreto, F.C. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]
- Sirrs, S.M.; Bichet, D.G.; Casey, R.; Clarke, J.T.R.; Lemoine, K.; Doucette, S.; West, M.L. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol. Genet. Metab. 2014, 111, 499–506. [Google Scholar] [CrossRef]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- Germain, D.P.; Fan, J.Q. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: In vitro and preclinical studies. Int. J. Clin. Pharmacol. Ther. 2009, 47, S111–S117. [Google Scholar] [PubMed]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Nicholls, K.; Giugliani, R.; Bichet, D.G.; Hughes, D.A.; Barisoni, L.M.; Colvin, R.B.; Jennette, J.C.; Skuban, N.; Castelli, J.P.; et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat-amenable variants: Data from the phase 3 randomized, multicenter, double-blind clinical trial and extension study. Genet. Med. 2019, 21, 1987–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashe, K.M.; Budman, E.; Bangari, D.S.; Siegel, C.S.; Nietupski, J.B.; Wang, B.; Desnick, R.J.; Scheule, R.K.; Leonard, J.P.; Cheng, S.H.; et al. Efficacy of enzyme and substrate reduction therapy with a novel antagonist of glucosylceramide synthase for fabry disease. Mol. Med. 2015, 21, 389–399. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Wood, K.M.; Rao, V.P.; Morin, J.; Bhamidipaty, S.; Labranche, T.P.; Gooch, R.L.; Bozal, F.; Bulawa, C.E.; Guild, B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016, 6, 20019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat-Andrés, Ó.; Pina-Buded, S.; Valverde-Navarro, A.A. Feasibility and diagnostic performance of including point-of-care ultrasound (POCUS) in preparticipation screening of young competitive athletes. Cardiol. Young 2020, 30, 1970–1972. [Google Scholar] [CrossRef]
- Fox, J.C.; Lahham, S.; Maldonado, G.; Klaus, S.; Aish, B.; Sylwanowicz, L.V.; Yanuck, J.; Wilson, S.P.; Shieh, M.; Anderson, C.L.; et al. Hypertrophic Cardiomyopathy in Youth Athletes: Successful Screening with Point-of-Care Ultrasound by Medical Students: Successful. J. Ultrasound Med. 2017, 36, 1109–1115. [Google Scholar] [CrossRef]
- Moulson, N.; Jaff, Z.; Wiltshire, V.; Taylor, T.; O’Connor, H.M.; Hopman, W.M.; Johri, A.M. Feasibility and Reliability of Nonexpert POCUS for Cardiovascular Preparticipation Screening of Varsity Athletes: The SHARP Protocol. Can. J. Cardiol. 2019, 35, 35–41. [Google Scholar] [CrossRef]
- Ko, W.Y.; Siontis, K.C.; Attia, Z.I.; Carter, R.E.; Kapa, S.; Ommen, S.R.; Demuth, S.J.; Ackerman, M.J.; Gersh, B.J.; Arruda-Olson, A.M.; et al. Detection of Hypertrophic Cardiomyopathy Using a Convolutional Neural Network-Enabled Electrocardiogram. J. Am. Coll. Cardiol. 2020, 75, 722–733. [Google Scholar] [CrossRef]
- Martini, N.; Aimo, A.; Barison, A.; Della Latta, D.; Vergaro, G.; Aquaro, G.D.; Ripoli, A.; Emdin, M.; Chiappino, D. Deep learning to diagnose cardiac amyloidosis from cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2020, 22. [Google Scholar] [CrossRef]
Figure 1.
Structural abnormalities detected on echocardiography in patients with Fabry cardiomyopathy. (A) Parasternal long-axis view shows increased left ventricular wall thickness (the hallmark feature of Fabry cardiomyopathy) along with thickening of the aortic and mitral valves as well as aortic dilatation. (B) Parasternal short-axis view shows prominent papillary muscle and binary sign (hyperechogenic endocardial layer of glycolipid-enriched smooth muscle cells adjacent to a hypoechogenic subendocardial layer relatively devoid of glycolipids). (C) Severe biatrial enlargement with a device lead seen in the right-sided chambers for treatment of advanced conduction disease. (D) Reduced longitudinal strain in the basal inferolateral segment characteristic of Fabry cardiomyopathy.
Figure 1.
Structural abnormalities detected on echocardiography in patients with Fabry cardiomyopathy. (A) Parasternal long-axis view shows increased left ventricular wall thickness (the hallmark feature of Fabry cardiomyopathy) along with thickening of the aortic and mitral valves as well as aortic dilatation. (B) Parasternal short-axis view shows prominent papillary muscle and binary sign (hyperechogenic endocardial layer of glycolipid-enriched smooth muscle cells adjacent to a hypoechogenic subendocardial layer relatively devoid of glycolipids). (C) Severe biatrial enlargement with a device lead seen in the right-sided chambers for treatment of advanced conduction disease. (D) Reduced longitudinal strain in the basal inferolateral segment characteristic of Fabry cardiomyopathy.


{kind=link}
{kind=link}
Table 1.
Cardiac manifestations of Fabry disease.
| Structural abnormalities detected by cardiac imaging |
|
| Electrophysiologic abnormalities detected by ECG or prolonged rhythm monitoring |
|
Abbreviations: AV, atrioventricular; ECG, electrocardiogram; LV, left ventricular; MRI, magnetic resonance imaging; RV, right ventricular; VT, ventricular tachycardia.
Table 2.
Differential diagnosis of increased LV wall thickness and common findings on patient history, ECG, echocardiography, and CMR.
Table 2.
Differential diagnosis of increased LV wall thickness and common findings on patient history, ECG, echocardiography, and CMR.
| Patient History | ECG | Echocardiography | CMR | |
|---|---|---|---|---|
| Fabry Cardiomyopathy |
|
|
|
|
| Hypertension |
|
|
|
|
| Athlete’s Heart |
|
|
|
|
| Aortic Stenosis |
|
|
|
|
| Hypertrophic Cardiomyopathy |
|
|
|
|
| Cardiac Amyloidosis |
|
|
|
|
Abbreviations: CMR, cardiovascular magnetic resonance imaging; ECG, electrocardiogram; GLS, global longitudinal strain; LGE, late gadolinium enhancement; LV, left ventricular; LVH, left ventricular hypertrophy, LVOT, left ventricular outflow tract; PM, papillary muscle; SVI, stroke volume index.
Table 3.
Comparison of approved and investigational disease-modifying therapies for Fabry disease.
| Disease-Modifying Therapy | Advantages | Disadvantages |
|---|---|---|
First-generation ERT
|
|
|
Oral chaperone therapy
|
|
|
Second-generation ERT
|
|
|
Substrate reduction therapy
|
|
|
| Gene therapy |
|
|
Table 4.
Management of the cardiovascular manifestations of Fabry disease.
| Structural abnormalities that can be present on cardiac imaging |
|
| Electrophysiologic abnormalities detected by ECG or rhythm monitoring |
|
| Other cardiovascular considerations in patients with Fabry disease |
|
Abbreviations: ACE, angiotensin converting enzyme; ARB, angiotensin II receptor blocker; AV, atrioventricular; ERT, enzyme replacement therapy; ICD, implantable cardioverter-defibrillator; LV, left ventricular; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yim, J.; Yau, O.; Yeung, D.F.; Tsang, T.S.M. Fabry Cardiomyopathy: Current Practice and Future Directions. Cells 2021, 10, 1532. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061532
AMA Style
Yim J, Yau O, Yeung DF, Tsang TSM. Fabry Cardiomyopathy: Current Practice and Future Directions. Cells. 2021; 10(6):1532. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061532
Chicago/Turabian StyleYim, Jeffrey, Olivia Yau, Darwin F. Yeung, and Teresa S. M. Tsang. 2021. "Fabry Cardiomyopathy: Current Practice and Future Directions" Cells 10, no. 6: 1532. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061532
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.