
Impact of Aldosterone on the Failing Myocardium: Insights from Mitochondria and Adrenergic Receptors Signaling and Function

 ,
,  and
and 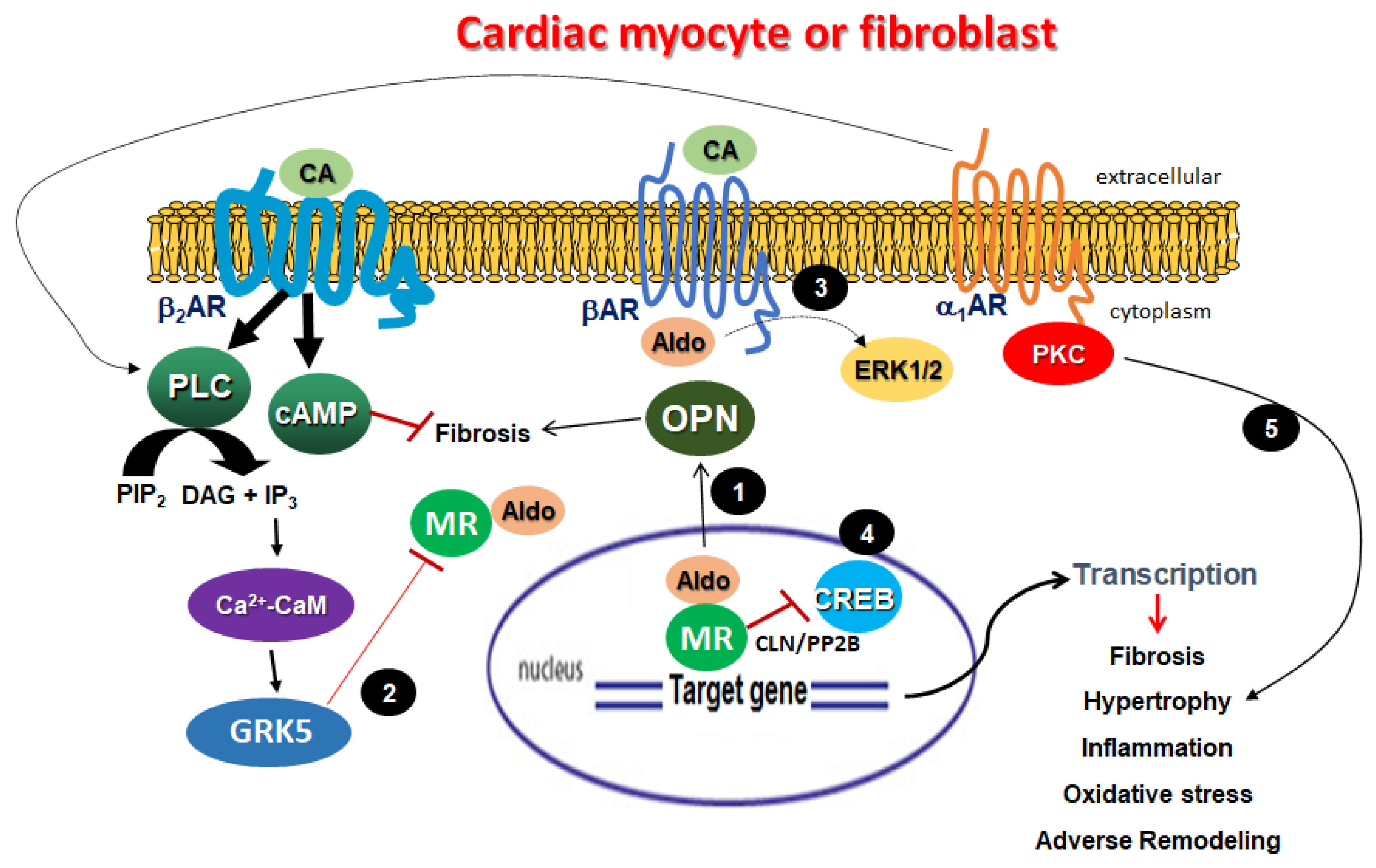{kind=link}
Abstract
:1. Introduction
2. Aldosterone and Mitochondrial Dysfunction in the Failing Heart
3. Cardiac Adrenergic Receptor Signaling and Dysregulation in HF
4. Aldosterone-Induced Adrenergic Receptor Dysfunction in the Failing Myocardium
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aldo | Aldosterone |
| Ca2+-CaM | Calcium-bound calmodulin |
| CA | Catecholamine |
| cAMP | Cyclic 3′,5′-adenosine monophosphate |
| CLN | Calcineurin |
| CREB | cAMP response element-binding protein |
| DAG | 2′-Diacylglycerol |
| ERK | Extracellular signal-regulate kinase |
| GRK5 | G protein-coupled receptor kinase-5 |
| IP3 | Inositol 1′,4′,5′-trisphopshate |
| OPN | Osteopontin |
| PIP2 | Phosphatidylinositol 4′,5′-bisphosphate |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PP2B | Protein phosphatase-2B |
References
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Parker, B.M.; Wertz, S.L.; Pollard, C.M.; Desimine, V.L.; Maning, J.; McCrink, K.A.; Lymperopoulos, A. Novel Insights into the Crosstalk between Mineralocorticoid Receptor and G Protein-Coupled Receptors in Heart Adverse Remodeling and Disease. Int. J. Mol. Sci. 2018, 19, 3764. [Google Scholar] [CrossRef] [PubMed]
- Parati, G.; Esler, M. The human sympathetic nervous system: Its relevance in hypertension and heart failure. Eur. Heart J. 2012, 33, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Quarti-Trevano, F.; Esler, M.D. Sympathetic activation in congestive heart failure: An updated overview. Heart Fail. Rev. 2021, 26, 173–182. [Google Scholar] [CrossRef]
- Cohn, J.N. Abnormalities of peripheral sympathetic nervous system control in congestive heart failure. Circulation 1990, 82, I59–I67. [Google Scholar]
- Solesio, M.E.; Saez-Atienzar, S.; Jordan, J.; Galindo, M.F. 3-Nitropropionic acid induces autophagy by forming mitochondrial permeability transition pores rather than activating the mitochondrial fission pathway. Br. J. Pharm. 2013, 168, 63–75. [Google Scholar] [CrossRef]
- Solesio, M.E.; Saez-Atienzar, S.; Jordan, J.; Galindo, M.F. Characterization of mitophagy in the 6-hydoxydopamine Parkinson’s disease model. Toxicol. Sci. 2012, 129, 411–420. [Google Scholar] [CrossRef]
- Solesio, M.E.; Prime, T.A.; Logan, A.; Murphy, M.P.; Del Mar Arroyo-Jimenez, M.; Jordan, J.; Galindo, M.F. The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim. Biophys. Acta 2013, 1832, 174–182. [Google Scholar] [CrossRef]
- Solesio, M.E.; Peixoto, P.M.; Debure, L.; Madamba, S.M.; de Leon, M.J.; Wisniewski, T.; Pavlov, E.V.; Fossati, S. Carbonic anhydrase inhibition selectively prevents amyloid β neurovascular mitochondrial toxicity. Aging Cell 2018, 17, e12787. [Google Scholar] [CrossRef]
- Liu, Z.; Solesio, M.E.; Schaffler, M.B.; Frikha-Benayed, D.; Rosen, C.J.; Werner, H.; Kopchick, J.J.; Pavlov, E.V.; Abramov, A.Y.; Yakar, S. Mitochondrial Function Is Compromised in Cortical Bone Osteocytes of Long-Lived Growth Hormone Receptor Null Mice. J. Bone Miner. Res. 2019, 34, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Galindo, M.F.; Solesio, M.E.; Atienzar-Aroca, S.; Zamora, M.J.; Jordan Bueso, J. Mitochondrial dynamics and mitophagy in the 6-hydroxydopamine preclinical model of Parkinson’s disease. Parkinsons Dis. 2012, 2012, 131058. [Google Scholar] [CrossRef]
- Borden, E.A.; Furey, M.; Gattone, N.J.; Hambardikar, V.D.; Liang, X.H.; Scoma, E.R.; Abou Samra, A.; LR, D.G.; Dennis, D.J.; Fricker, D.; et al. Is there a link between inorganic polyphosphate (polyP), mitochondria, and neurodegeneration? Pharm. Res. 2020, 105211. [Google Scholar] [CrossRef]
- Baltanas, A.; Solesio, M.E.; Zalba, G.; Galindo, M.F.; Fortuno, A.; Jordan, J. The senescence-accelerated mouse prone-8 (SAM-P8) oxidative stress is associated with upregulation of renal NADPH oxidase system. J. Physiol. Biochem. 2013, 69, 927–935. [Google Scholar] [CrossRef]
- Solesio, M.E.; Mitaishvili, E.; Lymperopoulos, A. Adrenal βarrestin1 targeting for tobacco-associated cardiac dysfunction treatment: Aldosterone production as the mechanistic link. Pharm. Res. Perspect. 2019, 7, e00497. [Google Scholar] [CrossRef]
- Miyata, K.; Rahman, M.; Shokoji, T.; Nagai, Y.; Zhang, G.X.; Sun, G.P.; Kimura, S.; Yukimura, T.; Kiyomoto, H.; Kohno, M.; et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J. Am. Soc. Nephrol. 2005, 16, 2906–2912. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kobara, M.; Abe, M.; Tanaka, N.; Gouda, E.; Toba, H.; Yamada, H.; Tatsumi, T.; Nakata, T.; Matsubara, H. Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens Res. 2008, 31, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, A.A.; Chen, L.; Malik, Z.A. Heart failure and mitochondrial dysfunction: The role of mitochondrial fission/fusion abnormalities and new therapeutic strategies. J. Cardiovasc. Pharm. 2014, 63, 196–206. [Google Scholar] [CrossRef]
- Villar, P.; Breton, B.; Garcia-Pavia, P.; Gonzalez-Paramos, C.; Blazquez, A.; Gomez-Bueno, M.; Garcia-Silva, T.; Garcia-Consuegra, I.; Martin, M.A.; Garesse, R.; et al. Cardiac dysfunction in mitochondrial disease. Clinical and molecular features. Circ. J. 2013, 77, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia--reperfusion, aging, and heart failure. J. Mol. Cell Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef]
- Ibarrola, J.; Sadaba, R.; Martinez-Martinez, E.; Garcia-Pena, A.; Arrieta, V.; Alvarez, V.; Fernandez-Celis, A.; Gainza, A.; Cachofeiro, V.; Santamaria, E.; et al. Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12. Sci. Rep. 2018, 8, 6801. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondrial dysfunction in heart failure. Heart Fail. Rev. 2013, 18, 607–622. [Google Scholar] [CrossRef]
- Tong, M.; Zablocki, D.; Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J. Mol. Cell Cardiol. 2020, 142, 138–145. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef]
- Yao, K.; Zhang, W.W.; Yao, L.; Yang, S.; Nie, W.; Huang, F. Carvedilol promotes mitochondrial biogenesis by regulating the PGC-1/TFAM pathway in human umbilical vein endothelial cells (HUVECs). Biochem. Biophys. Res. Commun. 2016, 470, 961–966. [Google Scholar] [CrossRef]
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Elustondo, P.A.; Zakharian, E.; Pavlov, E.V. Inorganic polyphosphate (polyP) as an activator and structural component of the mitochondrial permeability transition pore. Biochem. Soc. Trans. 2016, 44, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Solesio, M.E.; Garcia Del Molino, L.C.; Elustondo, P.A.; Diao, C.; Chang, J.C.; Pavlov, E.V. Inorganic polyphosphate is required for sustained free mitochondrial calcium elevation, following calcium uptake. Cell Calcium 2020, 86, 102127. [Google Scholar] [CrossRef]
- Solesio, M.E.; Xie, L.; McIntyre, B.; Ellenberger, M.; Mitaishvili, E.; Bhadra-Lobo, S.; Bettcher, L.F.; Bazil, J.N.; Raftery, D.; Jakob, U.; et al. Depletion of mitochondrial inorganic polyphosphate (polyP) in mammalian cells causes metabolic shift from oxidative phosphorylation to glycolysis. Biochem. J. 2021, 478, 1631–1646. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Gomez-Garcia, M.R.; Shiba, T.; Porter, G.A., Jr.; Pavlov, E.V.; Bers, D.M.; Dedkova, E.N. Dual role of inorganic polyphosphate in cardiac myocytes: The importance of polyP chain length for energy metabolism and mPTP activation. Arch. Biochem. Biophys. 2019, 662, 177–189. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Gomez-Garcia, M.R.; Blatter, L.A.; Pavlov, E.; Dedkova, E.N. Inorganic polyphosphate is a potent activator of the mitochondrial permeability transition pore in cardiac myocytes. J. Gen. Physiol. 2012, 139, 321–331. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Blatter, L.A.; Pavlov, E.; Dedkova, E.N. Inorganic polyphosphate--an unusual suspect of the mitochondrial permeability transition mystery. Channels 2012, 6, 463–467. [Google Scholar] [CrossRef]
- Chen, D.; Li, X.; Zhang, L.; Zhu, M.; Gao, L. A high-fat diet impairs mitochondrial biogenesis, mitochondrial dynamics, and the respiratory chain complex in rat myocardial tissues. J. Cell. Biochem. 2018, 119, 9602. [Google Scholar] [CrossRef]
- Birner, C.; Dietl, A.; Deutzmann, R.; Schröder, J.; Schmid, P.; Jungbauer, C.; Resch, M.; Endemann, D.; Stark, K.; Riegger, G. Proteomic profiling implies mitochondrial dysfunction in tachycardia-induced heart failure. J. Card. Fail. 2012, 18, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Dabkowski, E.R.; Baseler, W.A.; Williamson, C.L.; Powell, M.; Razunguzwa, T.T.; Frisbee, J.C.; Hollander, J.M. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H529–H540. [Google Scholar] [CrossRef]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation 2021, 143, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Sheeran, F.L.; Pepe, S. Mitochondrial bioenergetics and dysfunction in failing heart. Mitochondrial Dyn. Cardiovasc. Med. 2017, 65–80. [Google Scholar]
- Kiyuna, L.A.; e Albuquerque, R.P.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Ribeiro Junior, R.F.; Dabkowski, E.R.; Shekar, K.C.; KA, O.C.; Hecker, P.A.; Murphy, M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018, 117, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Bollag, W.B. Regulation of aldosterone synthesis and secretion. Compr. Physiol. 2014, 4, 1017–1055. [Google Scholar] [PubMed]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar]
- Alhayek, S.; Preuss, C.V. Beta 1 Receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/30422499/ (accessed on 3 May 2021).
- De Giusti, V.C.; Caldiz, C.I.; Ennis, I.L.; Perez, N.G.; Cingolani, H.E.; Aiello, E.A. Mitochondrial reactive oxygen species (ROS) as signaling molecules of intracellular pathways triggered by the cardiac renin-angiotensin II-aldosterone system (RAAS). Front. Physiol. 2013, 4, 126. [Google Scholar] [CrossRef] [PubMed]
- Daubert, M.A.; Yow, E.; Dunn, G.; Marchev, S.; Barnhart, H.; Douglas, P.S.; O’Connor, C.; Goldstein, S.; Udelson, J.E.; Sabbah, H.N. Novel Mitochondria-Targeting Peptide in Heart Failure Treatment: A Randomized, Placebo-Controlled Trial of Elamipretide. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Koyanagi, T.; Palaniyandi, S.S.; Fajardo, G.; Churchill, E.N.; Budas, G.; Disatnik, M.H.; Bernstein, D.; Brum, P.C.; Mochly-Rosen, D. Pharmacological inhibition of βIIPKC is cardioprotective in late-stage hypertrophy. J. Mol. Cell Cardiol. 2011, 51, 980–987. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Boer, B.N.; Grinberg, M.; Brum, P.C.; Mochly-Rosen, D. Protein quality control disruption by PKCβII in heart failure; rescue by the selective PKCβII inhibitor, βIIV5-3. PLoS ONE 2012, 7, e33175. [Google Scholar] [CrossRef]
- Budas, G.; Costa, H.M., Jr.; Ferreira, J.C.; Teixeira da Silva Ferreira, A.; Perales, J.; Krieger, J.E.; Mochly-Rosen, D.; Schechtman, D. Identification of epsilonPKC targets during cardiac ischemic injury. Circ. J. 2012, 76, 1476–1485. [Google Scholar] [CrossRef]
- Churchill, E.N.; Ferreira, J.C.; Brum, P.C.; Szweda, L.I.; Mochly-Rosen, D. Ischaemic preconditioning improves proteasomal activity and increases the degradation of deltaPKC during reperfusion. Cardiovasc. Res. 2010, 85, 385–394. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Green, D.R.; Van Houten, B. SnapShot: Mitochondrial quality control. Cell 2011, 147, 950.e1. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.P.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef]
- Fountain, J.H.; Lappin, S.L. Physiology, Renin Angiotensin System. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/29261862/ (accessed on 3 May 2021).
- Grossmann, C.; Krug, A.W.; Freudinger, R.; Mildenberger, S.; Voelker, K.; Gekle, M. Aldosterone-induced EGFR expression: Interaction between the human mineralocorticoid receptor and the human EGFR promoter. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1790–E1800. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kawai, T.; O’Brien, S.; Thomas, W.; Harris, R.C.; Eguchi, S. Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu. Rev. Pharm. Toxicol. 2016, 56, 627–653. [Google Scholar] [CrossRef]
- Aroor, A.R.; Demarco, V.G.; Jia, G.; Sun, Z.; Nistala, R.; Meininger, G.A.; Sowers, J.R. The role of tissue Renin-Angiotensin-aldosterone system in the development of endothelial dysfunction and arterial stiffness. Front. Endocrinol. 2013, 4, 161. [Google Scholar] [CrossRef]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Long, J.; Liu, J. Hyperglycemia-Associated Oxidative Stress Induces Autophagy: Involvement of the ROS-ERK/JNK-p53 Pathway. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2014; pp. 105–115. [Google Scholar]
- Lefranc, C.; Friederich-Persson, M.; Braud, L.; Palacios-Ramirez, R.; Karlsson, S.; Boujardine, N.; Motterlini, R.; Jaisser, F.; Nguyen Dinh Cat, A. MR (Mineralocorticoid Receptor) Induces Adipose Tissue Senescence and Mitochondrial Dysfunction Leading to Vascular Dysfunction in Obesity. Hypertension 2019, 73, 458–468. [Google Scholar] [CrossRef]
- Diviani, D.; Osman, H.; Delaunay, M.; Kaiser, S. The role of A-kinase anchoring proteins in cardiac oxidative stress. Biochem. Soc. Trans. 2019, 47, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Perrino, C.; Feliciello, A.; Schiattarella, G.G.; Esposito, G.; Guerriero, R.; Zaccaro, L.; Del Gatto, A.; Saviano, M.; Garbi, C.; Carangi, R.; et al. AKAP121 downregulation impairs protective cAMP signals, promotes mitochondrial dysfunction, and increases oxidative stress. Cardiovasc. Res. 2010, 88, 101–110. [Google Scholar] [CrossRef]
- Schmitz, B.; Brand, S.M.; Brand, E. Aldosterone signaling and soluble adenylyl cyclase-a nexus for the kidney and vascular endothelium. Biochim. Biophys. Acta 2014, 1842, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Spat, A.; Szanda, G. Mitochondrial cAMP and Ca2+ metabolism in adrenocortical cells. Pflügers Arch. 2018, 470, 1141–1148. [Google Scholar] [CrossRef]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef]
- Reiken, S.; Lacampagne, A.; Zhou, H.; Kherani, A.; Lehnart, S.E.; Ward, C.; Huang, F.; Gaburjakova, M.; Gaburjakova, J.; Rosemblit, N.; et al. PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: Defective regulation in heart failure. J. Cell Biol. 2003, 160, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef]
- Bhayana, V.; Alto, L.E.; Dhalla, N.S. The effects of β-adrenergic receptor blockers on heart mitochondrial metabolism. Gen. Pharm. 1980, 11, 271–274. [Google Scholar] [CrossRef]
- Panchal, A.R.; Stanley, W.C.; Kerner, J.; Sabbah, H.N. Beta-receptor blockade decreases carnitine palmitoyl transferase I activity in dogs with heart failure. J. Card. Fail. 1998, 4, 121–126. [Google Scholar] [CrossRef]
- Gómez, A.; Sánchez-Roman, I.; Gomez, J.; Cruces, J.; Mate, I.; Lopez-Torres, M.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; De la Fuente, M. Lifelong treatment with atenolol decreases membrane fatty acid unsaturation and oxidative stress in heart and skeletal muscle mitochondria and improves immunity and behavior, without changing mice longevity. Aging Cell 2014, 13, 551–560. [Google Scholar] [CrossRef]
- Zugck, C.; Haunstetter, A.; Kruger, C.; Kell, R.; Schellberg, D.; Kubler, W.; Haass, M. Impact of beta-blocker treatment on the prognostic value of currently used risk predictors in congestive heart failure. J. Am. Coll. Cardiol. 2002, 39, 1615–1622. [Google Scholar] [CrossRef]
- O’Neill, J.O.; Young, J.B.; Pothier, C.E.; Lauer, M.S. Peak oxygen consumption as a predictor of death in patients with heart failure receiving β-blockers. Circulation 2005, 111, 2313–2318. [Google Scholar] [CrossRef]
- Armour, J.A.; Murphy, D.A.; Yuan, B.X.; Macdonald, S.; Hopkins, D.A. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat. Rec. 1997, 247, 289–298. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Capote, L.A.; Mendez Perez, R.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur J. Pharm. 2015, 763, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A. Arrestins in the Cardiovascular System: An Update. Prog. Mol. Biol. Transl. Sci. 2018, 159, 27–57. [Google Scholar]
- Yoshikawa, T.; Port, J.D.; Asano, K.; Chidiak, P.; Bouvier, M.; Dutcher, D.; Roden, R.L.; Minobe, W.; Tremmel, K.D.; Bristow, M.R. Cardiac adrenergic receptor effects of carvedilol. Eur. Heart J. 1996, 17 (Suppl. B), 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Shannon, T.R. Calcium movements inside the sarcoplasmic reticulum of cardiac myocytes. J. Mol. Cell Cardiol. 2013, 58, 59–66. [Google Scholar] [CrossRef]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of β1- and β2-adrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef]
- Chesley, A.; Lundberg, M.S.; Asai, T.; Xiao, R.P.; Ohtani, S.; Lakatta, E.G.; Crow, M.T. The β2-adrenergic receptor delivers an antiapoptotic signal to cardiac myocytes through Gi-dependent coupling to phosphatidylinositol 3′-kinase. Circ. Res. 2000, 87, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Cora, N.; Maning, J.; Brill, A.R.; Sizova, A. Signaling and function of cardiac autonomic nervous system receptors: Insights from the GPCR signalling universe. FEBS J. 2021, 288, 2645–2659. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Levine, T.B.; Olivari, M.T.; Garberg, V.; Lura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N. Engl. J. Med. 1984, 311, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Kingwell, B.A.; Thompson, J.M.; Kaye, D.M.; McPherson, G.A.; Jennings, G.L.; Esler, M.D. Heart rate spectral analysis, cardiac norepinephrine spillover, and muscle sympathetic nerve activity during human sympathetic nervous activation and failure. Circulation 1994, 90, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Li, H.; Wang, J.J.; Zhang, J.S.; Shen, J.; An, X.B.; Zhang, C.C.; Wu, J.M.; Song, Y.; Wang, X.Y.; et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart J. 2018, 39, 60–69. [Google Scholar] [CrossRef]
- Zhao, W.B.; Lu, Q.; Nguyen, M.N.; Su, Y.; Ziemann, M.; Wang, L.N.; Kiriazis, H.; Puthalakath, H.; Sadoshima, J.; Hu, H.Y.; et al. Stimulation of β-adrenoceptors up-regulates cardiac expression of galectin-3 and BIM through the Hippo signalling pathway. Br. J. Pharm. 2019, 176, 2465–2481. [Google Scholar] [CrossRef]
- Du, X.J.; Gao, X.M.; Wang, B.; Jennings, G.L.; Woodcock, E.A.; Dart, A.M. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic receptors in the heart. Cardiovasc. Res. 2000, 48, 448–454. [Google Scholar] [CrossRef]
- Engelhardt, S.; Hein, L.; Wiesmann, F.; Lohse, M.J. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7059–7064. [Google Scholar] [CrossRef]
- Liggett, S.B.; Tepe, N.M.; Lorenz, J.N.; Canning, A.M.; Jantz, T.D.; Mitarai, S.; Yatani, A.; Dorn, G.W., 2nd. Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: Critical role for expression level. Circulation 2000, 101, 1707–1714. [Google Scholar] [CrossRef]
- Cleland, J.G.F.; Bunting, K.V.; Flather, M.D.; Altman, D.G.; Holmes, J.; Coats, A.J.S.; Manzano, L.; McMurray, J.J.V.; Ruschitzka, F.; van Veldhuisen, D.J.; et al. Beta-blockers in Heart Failure Collaborative, G. Beta-blockers for heart failure with reduced, mid-range, and preserved ejection fraction: An individual patient-level analysis of double-blind randomized trials. Eur. Heart J. 2018, 39, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Lechat, P.; Packer, M.; Chalon, S.; Cucherat, M.; Arab, T.; Boissel, J.P. Clinical effects of β-adrenergic blockade in chronic heart failure: A meta-analysis of double-blind, placebo-controlled, randomized trials. Circulation 1998, 98, 1184–1191. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by β-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Peter, P.S.; Brady, J.E.; Yan, L.; Chen, W.; Engelhardt, S.; Wang, Y.; Sadoshima, J.; Vatner, S.F.; Vatner, D.E. Inhibition of p38α MAPK rescues cardiomyopathy induced by overexpressed β2-adrenergic receptor, but not β1-adrenergic receptor. J. Clin. Investig. 2007, 117, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Grisanti, L.A.; Schumacher, S.M.; Tilley, D.G.; Koch, W.J. Designer Approaches for G Protein-Coupled Receptor Modulation for Cardiovascular Disease. JACC Basic Transl. Sci. 2018, 3, 550–562. [Google Scholar] [CrossRef]
- Conner, D.A.; Mathier, M.A.; Mortensen, R.M.; Christe, M.; Vatner, S.F.; Seidman, C.E.; Seidman, J.G. β-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to β-adrenergic stimulation. Circ. Res. 1997, 81, 1021–1026. [Google Scholar] [CrossRef]
- Watari, K.; Nakaya, M.; Nishida, M.; Kim, K.M.; Kurose, H. β-arrestin2 in infiltrated macrophages inhibits excessive inflammation after myocardial infarction. PLoS ONE 2013, 8, e68351. [Google Scholar] [CrossRef]
- Bathgate-Siryk, A.; Dabul, S.; Pandya, K.; Walklett, K.; Rengo, G.; Cannavo, A.; De Lucia, C.; Liccardo, D.; Gao, E.; Leosco, D.; et al. Negative impact of β-arrestin-1 on post-myocardial infarction heart failure via cardiac and adrenal-dependent neurohormonal mechanisms. Hypertension 2014, 63, 404–412. [Google Scholar] [CrossRef] [PubMed]
- McCrink, K.A.; Maning, J.; Vu, A.; Jafferjee, M.; Marrero, C.; Brill, A.; Bathgate-Siryk, A.; Dabul, S.; Koch, W.J.; Lymperopoulos, A. β-Arrestin2 Improves Post-Myocardial Infarction Heart Failure via Sarco(endo)plasmic Reticulum Ca2+-ATPase-Dependent Positive Inotropy in Cardiomyocytes. Hypertension 2017, 70, 972–981. [Google Scholar] [CrossRef]
- McCrink, K.A.; Maning, J.; Vu, A.; Jafferjee, M.; Marrero, C.; Brill, A.; Bathgate-Siryk, A.; Dabul, S.; Koch, W.J.; Lymperopoulos, A. Cardiac βarrestin2 Improves Contractility and Adverse Remodeling in Heart Failure, But Is Underexpressed in Humans. J. Am. Coll. Cardiol. 2017, 70, 2948–2949. [Google Scholar] [CrossRef] [PubMed]
- Bristow, M.R.; Hershberger, R.E.; Port, J.D.; Gilbert, E.M.; Sandoval, A.; Rasmussen, R.; Cates, A.E.; Feldman, A.M. Beta-adrenergic pathways in nonfailing and failing human ventricular myocardium. Circulation 1990, 82, I12–I25. [Google Scholar]
- Bohm, M.; Flesch, M.; Schnabel, P. Role of G-proteins in altered β-adrenergic responsiveness in the failing and hypertrophied myocardium. Basic Res. Cardiol. 1996, 91 (Suppl. 2), 47–51. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Friberg, P.; Rundqvist, B.; Quyyumi, A.A.; Lambert, G.; Kaye, D.M.; Kopin, I.J.; Goldstein, D.S.; Esler, M.D. Cardiac sympathetic nerve function in congestive heart failure. Circulation 1996, 93, 1667–1676. [Google Scholar] [CrossRef]
- Bristow, M.R. Mechanism of action of beta-blocking agents in heart failure. Am. J. Cardiol. 1997, 80, 26L–40L. [Google Scholar] [CrossRef]
- Nikolaev, V.O.; Moshkov, A.; Lyon, A.R.; Miragoli, M.; Novak, P.; Paur, H.; Lohse, M.J.; Korchev, Y.E.; Harding, S.E.; Gorelik, J. β2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 2010, 327, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Garcia, D.; Walklett, K. Pharmacogenetics of cardiac inotropy. Pharmacogenomics 2014, 15, 1807–1821. [Google Scholar] [CrossRef]
- Xiao, R.P.; Zhang, S.J.; Chakir, K.; Avdonin, P.; Zhu, W.; Bond, R.A.; Balke, C.W.; Lakatta, E.G.; Cheng, H. Enhanced Gi signaling selectively negates β2-adrenergic receptor (AR)--but not β1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation 2003, 108, 1633–1639. [Google Scholar] [CrossRef]
- Zhu, W.; Petrashevskaya, N.; Ren, S.; Zhao, A.; Chakir, K.; Gao, E.; Chuprun, J.K.; Wang, Y.; Talan, M.; Dorn, G.W., 2nd; et al. Gi-biased β2AR signaling links GRK2 upregulation to heart failure. Circ. Res. 2012, 110, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.C.; Vallejos, X.; Siryk, A.; Rengo, G.; Cannavo, A.; Liccardo, D.; De Lucia, C.; Gao, E.; Leosco, D.; Koch, W.J.; et al. GRK2 blockade with βARKct is essential for cardiac β2-adrenergic receptor signaling towards increased contractility. Cell Commun. Signal. 2013, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Bohm, M.; Elce, J.S.; Erdmann, E.; Lohse, M.J. Altered expression of beta-adrenergic receptor kinase and beta 1-adrenergic receptors in the failing human heart. Circulation 1993, 87, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Funakoshi, H.; Eckhart, A.D.; Koch, W.J. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat. Med. 2007, 13, 315–323. [Google Scholar] [CrossRef]
- Du, X.J.; Autelitano, D.J.; Dilley, R.J.; Wang, B.; Dart, A.M.; Woodcock, E.A. β2-adrenergic receptor overexpression exacerbates development of heart failure after aortic stenosis. Circulation 2000, 101, 71–77. [Google Scholar] [CrossRef]
- Lee, G.J.; Yan, L.; Vatner, D.E.; Vatner, S.F. Mst1 inhibition rescues β1-adrenergic cardiomyopathy by reducing myocyte necrosis and non-myocyte apoptosis rather than myocyte apoptosis. Basic Res. Cardiol. 2015, 110, 7. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ge, Y.; Shi, Z.; Duan, X.; Wang, L.; Sun, X.; Zhang, K. Response inhibition in adolescent earthquake survivors with and without posttraumatic stress disorder: A combined behavioral and ERP study. Neurosci. Lett. 2010, 486, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, J.; Wright, P.T.; Lyon, A.R.; Harding, S.E. Spatial control of the βAR system in heart failure: The transverse tubule and beyond. Cardiovasc. Res. 2013, 98, 216–224. [Google Scholar] [CrossRef]
- Siryk-Bathgate, A.; Dabul, S.; Lymperopoulos, A. Current and future G protein-coupled receptor signaling targets for heart failure therapy. Drug Des. Dev. Ther. 2013, 7, 1209–1222. [Google Scholar]
- Xu, Q.; Dalic, A.; Fang, L.; Kiriazis, H.; Ritchie, R.H.; Sim, K.; Gao, X.M.; Drummond, G.; Sarwar, M.; Zhang, Y.Y.; et al. Myocardial oxidative stress contributes to transgenic β2-adrenoceptor activation-induced cardiomyopathy and heart failure. Br. J. Pharm. 2011, 162, 1012–1028. [Google Scholar] [CrossRef]
- Sheridan, D.J.; Autelitano, D.J.; Wang, B.; Percy, E.; Woodcock, E.A.; Du, X.J. β2-adrenergic receptor overexpression driven by α-MHC promoter is downregulated in hypertrophied and failing myocardium. Cardiovasc. Res. 2000, 47, 133–141. [Google Scholar] [CrossRef]
- Du, X.J.; Gao, X.M.; Jennings, G.L.; Dart, A.M.; Woodcock, E.A. Preserved ventricular contractility in infarcted mouse heart overexpressing β2-adrenergic receptors. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2456–H2463. [Google Scholar] [CrossRef]
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: A new model of Takotsubo cardiomyopathy. Circulation 2012, 126, 697–706. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Bouvet, M.; Blondeau, J.P.; Lezoualc’h, F. The Epac1 Protein: Pharmacological Modulators, Cardiac Signalosome and Pathophysiology. Cells 2019, 8, 1543. [Google Scholar] [CrossRef] [PubMed]
- Fazal, L.; Laudette, M.; Paula-Gomes, S.; Pons, S.; Conte, C.; Tortosa, F.; Sicard, P.; Sainte-Marie, Y.; Bisserier, M.; Lairez, O.; et al. Multifunctional Mitochondrial Epac1 Controls Myocardial Cell Death. Circ. Res. 2017, 120, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Szanda, G.; Wisniewski, E.; Rajki, A.; Spat, A. Mitochondrial cAMP exerts positive feedback on mitochondrial Ca2+ uptake via the recruitment of Epac1. J. Cell Sci. 2018, 131. [Google Scholar]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Okumura, S.; Fujita, T.; Cai, W.; Jin, M.; Namekata, I.; Mototani, Y.; Jin, H.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig. 2014, 124, 2785–2801. [Google Scholar] [CrossRef] [PubMed]
- Laurent, A.C.; Bisserier, M.; Lucas, A.; Tortosa, F.; Roumieux, M.; De Regibus, A.; Swiader, A.; Sainte-Marie, Y.; Heymes, C.; Vindis, C.; et al. Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 105, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Laudette, M.; Coluccia, A.; Sainte-Marie, Y.; Solari, A.; Fazal, L.; Sicard, P.; Silvestri, R.; Mialet-Perez, J.; Pons, S.; Ghaleh, B.; et al. Identification of a pharmacological inhibitor of Epac1 that protects the heart against acute and chronic models of cardiac stress. Cardiovasc. Res. 2019, 115, 1766–1777. [Google Scholar] [CrossRef]
- Wang, J.; Hanada, K.; Staus, D.P.; Makara, M.A.; Dahal, G.R.; Chen, Q.; Ahles, A.; Engelhardt, S.; Rockman, H.A. Galphai is required for carvedilol-induced β1 adrenergic receptor β-arrestin biased signaling. Nat. Commun. 2017, 8, 1706. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; West, T.M.; Liu, Y.; Reddy, G.R.; Barbagallo, F.; Xu, B.; Shi, Q.; Deng, B.; Wei, W.; et al. Carvedilol induces biased β1 adrenergic receptor-Nitric oxide synthase 3-cyclic guanylyl monophosphate signaling to promote cardiac contractility. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Littmann, T.; Gottle, M.; Reinartz, M.T.; Kalble, S.; Wainer, I.W.; Ozawa, T.; Seifert, R. Recruitment of β-arrestin 1 and 2 to the β2-adrenoceptor: Analysis of 65 ligands. J. Pharm. Exp. 2015, 355, 183–190. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Kawakami, K.; Aoki, J.; Messer, K.; Sunahara, R.; et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Desimine, V.L.; McCrink, K.A.; Parker, B.M.; Wertz, S.L.; Maning, J.; Lymperopoulos, A. Biased Agonism/Antagonism of Cardiovascular GPCRs for Heart Failure Therapy. Int. Rev. Cell Mol. Biol. 2018, 339, 41–61. [Google Scholar] [PubMed]
- Yang, H.Q.; Wang, L.P.; Gong, Y.Y.; Fan, X.X.; Zhu, S.Y.; Wang, X.T.; Wang, Y.P.; Li, L.L.; Xing, X.; Liu, X.X.; et al. β2-Adrenergic Stimulation Compartmentalizes β1 Signaling Into Nanoscale Local Domains by Targeting the C-Terminus of β1-Adrenoceptors. Circ. Res. 2019, 124, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Grisan, F.; Burdyga, A.; Iannucci, L.F.; Surdo, N.C.; Pozzan, T.; Di Benedetto, G.; Lefkimmiatis, K. Studying β1 and β2 adrenergic receptor signals in cardiac cells using FRET-based sensors. Prog. Biophys. Mol. Biol. 2020, 154, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, M.; Osman, H.; Kaiser, S.; Diviani, D. The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells 2019, 9, 69. [Google Scholar] [CrossRef]
- Turner, N.A.; Porter, K.E.; Smith, W.H.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic β2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef]
- Chen, C.; Du, J.; Feng, W.; Song, Y.; Lu, Z.; Xu, M.; Li, Z.; Zhang, Y. β-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCdelta/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br. J. Pharm. 2012, 166, 676–688. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Valiente-Alandi, I.; Nieman, M.L.; Sargent, M.A.; Lorenz, J.N.; Molkentin, J.D.; Blaxall, B.C. Pharmacological and Activated Fibroblast Targeting of Gβγ-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression. J. Am. Coll. Cardiol. 2017, 70, 958–971. [Google Scholar] [CrossRef]
- Yin, F.; Wang, Y.Y.; Du, J.H.; Li, C.; Lu, Z.Z.; Han, C.; Zhang, Y.Y. Noncanonical cAMP pathway and p38 MAPK mediate β2-adrenergic receptor-induced IL-6 production in neonatal mouse cardiac fibroblasts. J. Mol. Cell Cardiol. 2006, 40, 384–393. [Google Scholar] [CrossRef]
- Kiriazis, H.; Wang, K.; Xu, Q.; Gao, X.M.; Ming, Z.; Su, Y.; Moore, X.L.; Lambert, G.; Gibbs, M.E.; Dart, A.M.; et al. Knockout of β1- and β2-adrenoceptors attenuates pressure overload-induced cardiac hypertrophy and fibrosis. Br. J. Pharm. 2008, 153, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharm. 2012, 166, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.M.; Desimine, V.L.; Wertz, S.L.; Perez, A.; Parker, B.M.; Maning, J.; McCrink, K.A.; Shehadeh, L.A.; Lymperopoulos, A. Deletion of Osteopontin Enhances β2-Adrenergic Receptor-Dependent Anti-Fibrotic Signaling in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 1396. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, E.A.; Du, X.J.; Reichelt, M.E.; Graham, R.M. Cardiac α1-adrenergic drive in pathological remodelling. Cardiovasc. Res. 2008, 77, 452–462. [Google Scholar] [CrossRef]
- Jensen, B.C.; O’Connell, T.D.; Simpson, P.C. Alpha-1-adrenergic receptors in heart failure: The adaptive arm of the cardiac response to chronic catecholamine stimulation. J. Cardiovasc. Pharm. 2014, 63, 291–301. [Google Scholar] [CrossRef]
- O’Connell, T.D.; Swigart, P.M.; Rodrigo, M.C.; Ishizaka, S.; Joho, S.; Turnbull, L.; Tecott, L.H.; Baker, A.J.; Foster, E.; Grossman, W.; et al. α1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J. Clin. Investig. 2006, 116, 1005–1015. [Google Scholar] [CrossRef]
- Wang, B.H.; Du, X.J.; Autelitano, D.J.; Milano, C.A.; Woodcock, E.A. Adverse effects of constitutively active α1B-adrenergic receptors after pressure overload in mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1079–H1086. [Google Scholar] [CrossRef]
- Huang, Y.; Wright, C.D.; Merkwan, C.L.; Baye, N.L.; Liang, Q.; Simpson, P.C.; O’Connell, T.D. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 2007, 115, 763–772. [Google Scholar] [CrossRef]
- Myagmar, B.E.; Ismaili, T.; Swigart, P.M.; Raghunathan, A.; Baker, A.J.; Sahdeo, S.; Blevitt, J.M.; Milla, M.E.; Simpson, P.C. Coupling to Gq Signaling Is Required for Cardioprotection by an Alpha-1A-Adrenergic Receptor Agonist. Circ. Res. 2019, 125, 699–706. [Google Scholar] [CrossRef]
- Shi, T.; Papay, R.S.; Perez, D.M. α1A-Adrenergic receptor prevents cardiac ischemic damage through PKCdelta/GLUT1/4-mediated glucose uptake. J. Recept. Signal Transduct. Res. 2016, 36, 261–270. [Google Scholar] [CrossRef]
- Lin, F.; Owens, W.A.; Chen, S.; Stevens, M.E.; Kesteven, S.; Arthur, J.F.; Woodcock, E.A.; Feneley, M.P.; Graham, R.M. Targeted α1A-adrenergic receptor overexpression induces enhanced cardiac contractility but not hypertrophy. Circ. Res. 2001, 89, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Beak, J.; Huang, W.; Parker, J.S.; Hicks, S.T.; Patterson, C.; Simpson, P.C.; Ma, A.; Jin, J.; Jensen, B.C. An Oral Selective Alpha-1A Adrenergic Receptor Agonist Prevents Doxorubicin Cardiotoxicity. JACC Basic Transl. Sci. 2017, 2, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, M.D.; Chan, T.; Swigart, P.M.; Myagmar, B.E.; Dash, R.; Simpson, P.C. An Alpha-1A Adrenergic Receptor Agonist Prevents Acute Doxorubicin Cardiomyopathy in Male Mice. PLoS ONE 2017, 12, e0168409. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.C.; Swigart, P.M.; De Marco, T.; Hoopes, C.; Simpson, P.C. α1-Adrenergic receptor subtypes in nonfailing and failing human myocardium. Circ. Heart Fail. 2009, 2, 654–663. [Google Scholar] [CrossRef]
- Janssen, P.M.L.; Canan, B.D.; Kilic, A.; Whitson, B.A.; Baker, A.J. Human Myocardium Has a Robust α1A-Subtype Adrenergic Receptor Inotropic Response. J. Cardiovasc. Pharm. 2018, 72, 136–142. [Google Scholar] [CrossRef]
- Yu, Z.Y.; Tan, J.C.; McMahon, A.C.; Iismaa, S.E.; Xiao, X.H.; Kesteven, S.H.; Reichelt, M.E.; Mohl, M.C.; Smith, N.J.; Fatkin, D.; et al. RhoA/ROCK signaling and pleiotropic α1A-adrenergic receptor regulation of cardiac contractility. PLoS ONE 2014, 9, e99024. [Google Scholar] [CrossRef]
- Mohl, M.C.; Iismaa, S.E.; Xiao, X.H.; Friedrich, O.; Wagner, S.; Nikolova-Krstevski, V.; Wu, J.; Yu, Z.Y.; Feneley, M.; Fatkin, D.; et al. Regulation of murine cardiac contractility by activation of α1A-adrenergic receptor-operated Ca2+ entry. Cardiovasc. Res. 2011, 91, 310–319. [Google Scholar] [CrossRef]
- Hein, L.; Altman, J.D.; Kobilka, B.K. Two functionally distinct α2-adrenergic receptors regulate sympathetic neurotransmission. Nature 1999, 402, 181–184. [Google Scholar] [CrossRef]
- Brede, M.; Wiesmann, F.; Jahns, R.; Hadamek, K.; Arnolt, C.; Neubauer, S.; Lohse, M.J.; Hein, L. Feedback inhibition of catecholamine release by two different α2-adrenoceptor subtypes prevents progression of heart failure. Circulation 2002, 106, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Brede, M.; Nagy, G.; Philipp, M.; Sorensen, J.B.; Lohse, M.J.; Hein, L. Differential control of adrenal and sympathetic catecholamine release by α2-adrenoceptor subtypes. Mol. Endocrinol. 2003, 17, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Brede, M.; Philipp, M.; Knaus, A.; Muthig, V.; Hein, L. α2-adrenergic receptor subtypes-novel functions uncovered in gene-targeted mouse models. Biol. Cell 2004, 96, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Gao, E.; Ebert, S.N.; Dorn, G.W., 2nd; Koch, W.J. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J. Biol. Chem. 2010, 285, 16378–16386. [Google Scholar] [CrossRef]
- Jafferjee, M.; Reyes Valero, T.; Marrero, C.; McCrink, K.A.; Brill, A.; Lymperopoulos, A. GRK2 Up-Regulation Creates a Positive Feedback Loop for Catecholamine Production in Chromaffin Cells. Mol. Endocrinol. 2016, 30, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Lother, A.; Hein, L.; Gilsbach, R. Chronic cardiac pressure overload induces adrenal medulla hypertrophy and increased catecholamine synthesis. Basic Res. Cardiol. 2011, 106, 591–602. [Google Scholar] [CrossRef]
- Nguyen, K.; Kassimatis, T.; Lymperopoulos, A. Impaired desensitization of a human polymorphic α2B-adrenergic receptor variant enhances its sympatho-inhibitory activity in chromaffin cells. Cell Commun. Signal. 2011, 9, 5. [Google Scholar] [CrossRef]
- Small, K.M.; Wagoner, L.E.; Levin, A.M.; Kardia, S.L.; Liggett, S.B. Synergistic polymorphisms of beta1- and alpha2C-adrenergic receptors and the risk of congestive heart failure. N. Engl. J. Med. 2002, 347, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Fiuzat, M.; Carson, P.E.; Anand, I.S.; Plehn, J.F.; Gottlieb, S.S.; Silver, M.A.; Lindenfeld, J.; Miller, A.B.; White, M.; et al. Combinatorial pharmacogenetic interactions of bucindolol and β1, α2C adrenergic receptor polymorphisms. PLoS ONE 2012, 7, e44324. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Aukszi, B. Angiotensin receptor blocker drugs and inhibition of adrenal β-arrestin-1-dependent aldosterone production: Implications for heart failure therapy. World J. Cardiol. 2017, 9, 200–206. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Gesty-Palmer, D. Beyond desensitization: Physiological relevance of arrestin-dependent signaling. Pharm. Rev. 2010, 62, 305–330. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Koch, W.J. Adrenal β-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J. Am. Coll. Cardiol. 2011, 57, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; Rengo, G.; Zincarelli, C.; Kim, J.; Soltys, S.; Koch, W.J. An adrenal β-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5825–5830. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.M.; Ghandour, J.; Cora, N.; Perez, A.; Parker, B.M.; Desimine, V.L.; Wertz, S.L.; Pereyra, J.M.; Ferraino, K.E.; Patel, J.J.; et al. GRK2-Mediated Crosstalk between β-Adrenergic and Angiotensin II Receptors Enhances Adrenocortical Aldosterone Production In Vitro and In Vivo. Int. J. Mol. Sci. 2020, 21, 574. [Google Scholar] [CrossRef] [PubMed]
- Cora, N.; Ghandour, J.; Pollard, C.M.; Desimine, V.L.; Ferraino, K.E.; Pereyra, J.M.; Valiente, R.; Lymperopoulos, A. Nicotine-induced adrenal β-arrestin1 upregulation mediates tobacco-related hyperaldosteronism leading to cardiac dysfunction. World J. Cardiol. 2020, 12, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Ferraino, K.E.; Cora, N.; Pollard, C.M.; Sizova, A.; Maning, J.; Lymperopoulos, A. Adrenal angiotensin II type 1 receptor biased signaling: The case for "biased" inverse agonism for effective aldosterone suppression. Cell Signal. 2021, 82, 109967. [Google Scholar] [CrossRef] [PubMed]
- Maning, J.; McCrink, K.A.; Pollard, C.M.; Desimine, V.L.; Ghandour, J.; Perez, A.; Cora, N.; Ferraino, K.E.; Parker, B.M.; Brill, A.R.; et al. Antagonistic Roles of GRK2 and GRK5 in Cardiac Aldosterone Signaling Reveal GRK5-Mediated Cardioprotection via Mineralocorticoid Receptor Inhibition. Int. J. Mol. Sci. 2020, 21, 2868. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Chuprun, J.K.; Schwartz, M.; Koch, W.J. The evolving impact of g protein-coupled receptor kinases in cardiac health and disease. Physiol. Rev. 2015, 95, 377–404. [Google Scholar] [CrossRef]
- Marzano, F.; Rapacciuolo, A.; Ferrara, N.; Rengo, G.; Koch, W.J.; Cannavo, A. Targeting GRK5 for Treating Chronic Degenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1920. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.C.; Eguchi, A.; Lieu, M.; Roy, R.; Barr, E.W.; Ibetti, J.; Lucchese, A.M.; Peluzzo, A.M.; Gresham, K.; Chuprun, J.K.; et al. A peptide of the N terminus of GRK5 attenuates pressure-overload hypertrophy and heart failure. Sci. Signal. 2021, 14. [Google Scholar] [CrossRef]
- Liggett, S.B.; Cresci, S.; Kelly, R.J.; Syed, F.M.; Matkovich, S.J.; Hahn, H.S.; Diwan, A.; Martini, J.S.; Sparks, L.; Parekh, R.R., 2nd; et al. A GRK5 polymorphism that inhibits β-adrenergic receptor signaling is protective in heart failure. Nat. Med. 2008, 14, 510–517. [Google Scholar] [CrossRef]
- Wu, J.H.; Zhang, L.; Fanaroff, A.C.; Cai, X.; Sharma, K.C.; Brian, L.; Exum, S.T.; Shenoy, S.K.; Peppel, K.; Freedman, N.J. G protein-coupled receptor kinase-5 attenuates atherosclerosis by regulating receptor tyrosine kinases and 7-transmembrane receptors. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 308–316. [Google Scholar] [CrossRef]
- Monto, F.; Oliver, E.; Vicente, D.; Rueda, J.; Aguero, J.; Almenar, L.; Ivorra, M.D.; Barettino, D.; D’Ocon, P. Different expression of adrenoceptors and GRKs in the human myocardium depends on heart failure etiology and correlates to clinical variables. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H368–H376. [Google Scholar] [CrossRef]
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFκB transcriptional activity by inducing nuclear accumulation of IκBα. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823. [Google Scholar] [CrossRef]
- de Lucia, C.; Grisanti, L.A.; Borghetti, G.; Piedepalumbo, M.; Ibetti, J.; Maria Lucchese, A.; Barr, E.W.; Roy, R.; Dedo Okyere, A.; Christine Murphy, H.; et al. GRK5 contributes to impaired cardiac function and immune cell recruitment in post-ischemic heart failure. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Eguchi, A.; Coleman, R.; Gresham, K.; Gao, E.; Ibetti, J.; Chuprun, J.K.; Koch, W.J. GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Beyett, T.S.; Fraley, A.E.; Labudde, E.; Patra, D.; Coleman, R.C.; Eguchi, A.; Glukhova, A.; Chen, Q.; Williams, R.M.; Koch, W.J.; et al. Perturbation of the interactions of calmodulin with GRK5 using a natural product chemical probe. Proc. Natl. Acad. Sci. USA 2019, 116, 15895–15900. [Google Scholar] [CrossRef] [PubMed]
- Pabbidi, M.R.; Ji, X.; Maxwell, J.T.; Mignery, G.A.; Samarel, A.M.; Lipsius, S.L. Inhibition of cAMP-Dependent PKA Activates β2-Adrenergic Receptor Stimulation of Cytosolic Phospholipase A2 via Raf-1/MEK/ERK and IP3-Dependent Ca2+ Signaling in Atrial Myocytes. PLoS ONE 2016, 11, e0168505. [Google Scholar] [CrossRef]
- Hori, Y.; Touei, D.; Saitoh, R.; Yamagishi, M.; Kanai, K.; Hoshi, F.; Itoh, N. The Aldosterone Receptor Antagonist Eplerenone Inhibits Isoproterenol-Induced Collagen-I and 11β-HSD1 Expression in Rat Cardiac Fibroblasts and the Left Ventricle. Biol. Pharm. Bull. 2017, 40, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Wuttke, M.; Ruhs, S.; Seiferth, A.; Mildenberger, S.; Rabe, S.; Schwerdt, G.; Gekle, M. Mineralocorticoid receptor inhibits CREB signaling by calcineurin activation. FASEB J. 2010, 24, 2010–2019. [Google Scholar] [CrossRef]
- Karkoulias, G.; McCrink, K.A.; Maning, J.; Pollard, C.M.; Desimine, V.L.; Patsouras, N.; Psallidopoulos, M.; Taraviras, S.; Lymperopoulos, A.; Flordellis, C. Sustained GRK2-dependent CREB activation is essential for α2-adrenergic receptor-induced PC12 neuronal differentiation. Cell Signal. 2020, 66, 109446. [Google Scholar] [CrossRef] [PubMed]
- Lister, K.; Autelitano, D.J.; Jenkins, A.; Hannan, R.D.; Sheppard, K.E. Cross talk between corticosteroids and alpha-adrenergic signalling augments cardiomyocyte hypertrophy: A possible role for SGK1. Cardiovasc. Res. 2006, 70, 555–565. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guitart-Mampel, M.; Urquiza, P.; Borges, J.I.; Lymperopoulos, A.; Solesio, M.E. Impact of Aldosterone on the Failing Myocardium: Insights from Mitochondria and Adrenergic Receptors Signaling and Function. Cells 2021, 10, 1552. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061552
Guitart-Mampel M, Urquiza P, Borges JI, Lymperopoulos A, Solesio ME. Impact of Aldosterone on the Failing Myocardium: Insights from Mitochondria and Adrenergic Receptors Signaling and Function. Cells. 2021; 10(6):1552. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061552
Chicago/Turabian StyleGuitart-Mampel, Mariona, Pedro Urquiza, Jordana I. Borges, Anastasios Lymperopoulos, and Maria E. Solesio. 2021. "Impact of Aldosterone on the Failing Myocardium: Insights from Mitochondria and Adrenergic Receptors Signaling and Function" Cells 10, no. 6: 1552. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10061552