
Effects of Mild Excitotoxic Stimulus on Mitochondria Ca2+ Handling in Hippocampal Cultures of a Mouse Model of Alzheimer’s Disease


Abstract
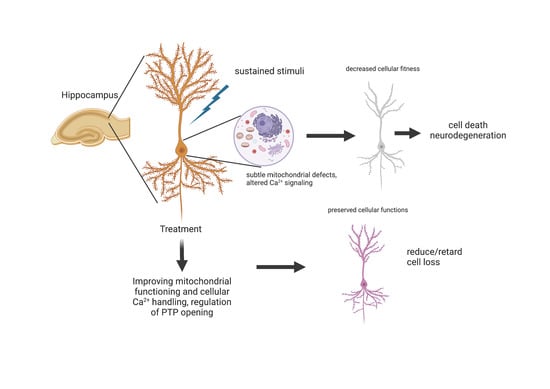
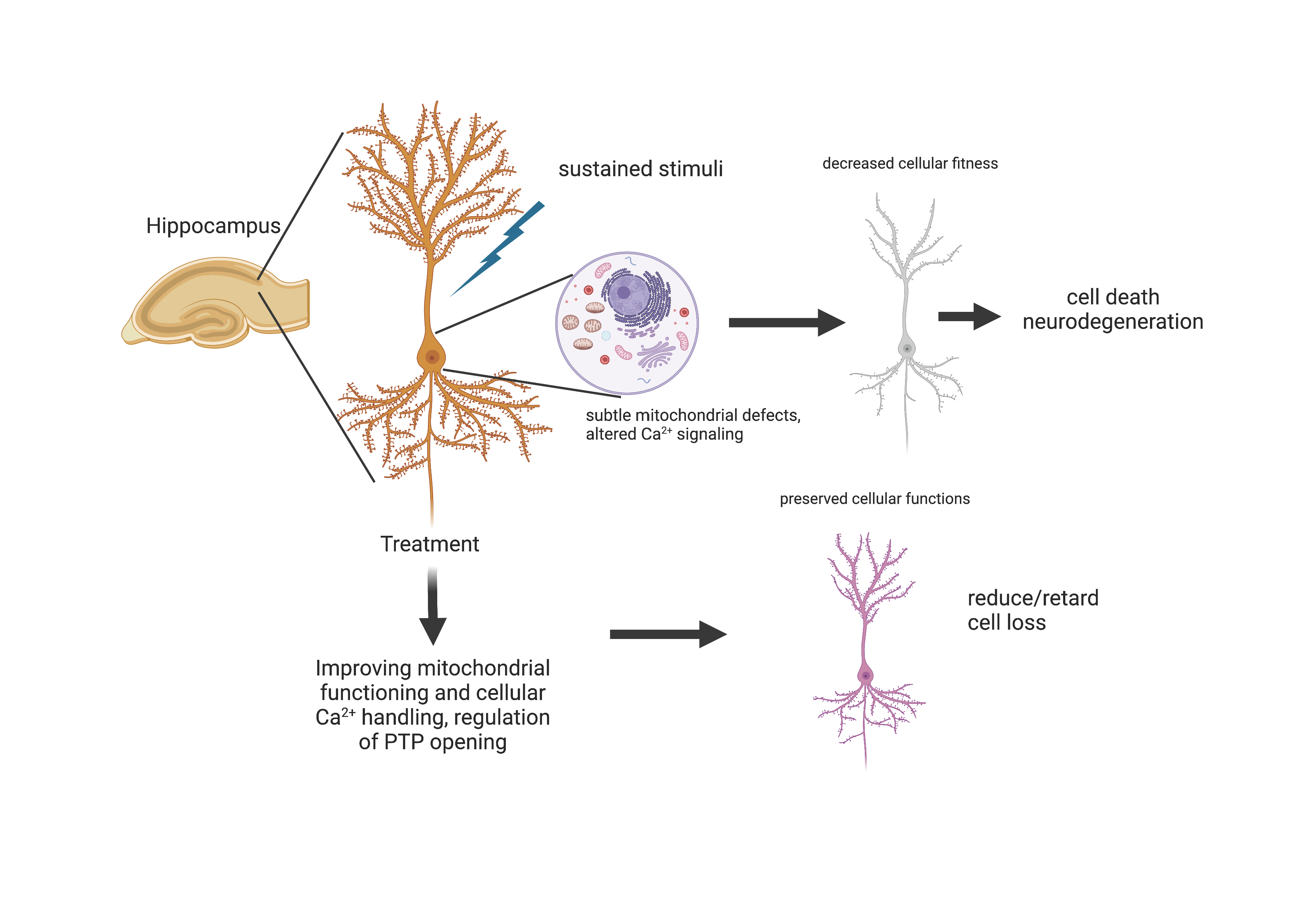
:
1. Introduction
2. Materials and Methods
2.1. Animal Handling and Care
2.2. Primary Neuronal Cultures
2.3. Adeno-Associated Virus (AAV) Production
2.4. Ca2+ Imaging
2.5. TMRM Experiment
2.6. Superoxide Measurements
2.7. H2O2 Measurements
2.8. Protein Extraction and Western Blotting
2.9. Immunofluorescence
3. Results
3.1. FAD Hippocampal Neurons Have Impaired Respiratory Capacity
3.2. FAD Hippocampal Neurons Are Less Able to Maintain a Stable Membrane Potential When Challenged with Inhibitors of the Respiratory Chain
3.3. Mitochondrial Reactive Oxygen Species Production
3.4. FAD Hippocampal Neurons Are More Sensitive to Glutamate Excitotoxicity
3.4.1. Effect of Glutamate on Cytosolic Ca2+ Transients
3.4.2. Effect of Glutamate on Mitochondrial Ca2+ Transients
3.5. Glutamate Excitotoxicity Impairs FAD Hippocampal Neurons Membrane Potential, Effect of Alisporivir
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermunt, L.; Sikkes, S.A.M.; Hout, A.; Handels, R.; Bos, I.; Flier, W.M.; Kern, S.; Ousset, P.; Maruff, P.; Skoog, I.; et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimer’s Dement. 2019, 15, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Altmann, A.; Ng, B.; Landau, S.M.; Jagust, W.J.; Greicius, M.D. Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 2015, 138, 3734–3746. [Google Scholar] [CrossRef]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2008, 6, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef] [PubMed]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef]
- Kish, S.J.; Mastrogiacomo, F.; Guttman, M.; Furukawa, Y.; Taanman, J.-W.; DožicDožic, S.; Pandolfo, M.; Lamarche, J.; DiStefano, L.; Chang, L.-J. Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: A nonspecific change? J. Neurochem. 1999, 72, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Arsic, N.; Zacchigna, S.; Zentilin, L.; Ramirez-Correa, G.; Pattarini, L.; Salvi, A.; Sinagra, G.; Giacca, M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004, 10, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Galla, L.; Gomiero, C.; Zentilin, L.; Giacca, M.; Giorgio, V.; Calì, T.; Pozzan, T.; Greotti, E.; Pizzo, P. Calcium signaling and mitochondrial function in presenilin 2 knock-out mice: Looking for any loss-of-function phenotype related to Alzheimer’s disease. Cells 2021, 10, 204. [Google Scholar] [CrossRef]
- Inagaki, K.; Fuess, S.; Storm, T.A.; Gibson, G.A.; Mctiernan, C.F.; Kay, M.A.; Nakai, H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006, 14, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, E.; Mingozzi, F.; Montane, J.; Leon, X.; Anguela, X.M.; Haurigot, V.; Edmonson, S.A.; Africa, L.; Zhou, S.; High, K.A.; et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010, 17, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zentilin, L.; Giacca, M. Competitive PCR for precise nucleic acid quantification. Nat. Protoc. 2007, 2, 2092–2104. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive red protein calcium indicators for imaging neural activity. eLife 2016, 5, e12727. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.E.; Giacomello, M.; Kortemme, T.; Hires, S.A.; Lev-Ram, V.; Baker, D.; Tsien, R.Y. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem. Biol. 2006, 13, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, D.R.; Gardiner, N.J. Glucose neurotoxicity. Nat. Rev. Neurosci. 2008, 9, 36–45. [Google Scholar] [CrossRef]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruas, J.S.; Siqueira-Santos, E.S.; Amigo, I.; Rodrigues-Silva, E.; Kowaltowski, A.J.; Castilho, R.F. Underestimation of the maximal capacity of the mitochondrial electron transport system in oligomycin-treated cells. PLoS ONE 2016, 11, e0150967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.L.; Watkins, F.H.; Steward, O.; Banker, G. Characterization of GABAergic neurons in hippocampal cell cultures. J. Neurocytol. 1994, 23, 279–295. [Google Scholar] [CrossRef]
- Pedersen, P.L.; Carafoli, E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem. Sci. 1987, 12, 146–150. [Google Scholar] [CrossRef]
- Arato-Oshima, T.; Matsui, H.; Wakizaka, A.; Homareda, H. Mechanism Responsible for Oligomycin-Induced Occlusion of Na+ within Na/K-ATPase*. Available online: https://0-reader-elsevier-com.brum.beds.ac.uk/reader/sd/pii/S0021925819783331?token=0F8A6B0F5E2E9CEB6893CBD67E210FC4F611C83A1B80DDDE8E14A8FB5CB21DD6179CA16812213BB9590235C387978DE7&originRegion=eu-west-1&originCreation=20210622170219 (accessed on 22 June 2021).
- Schägger, H.; Ohm, T.G. Human diseases with defects in oxidative phosphorylation: 2. F1F0 ATP-synthase defects in Alzheimer disease revealed by blue native polyacrylamide gel electrophoresis. Eur. J. Biochem. 1995, 227, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S.; et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 2016, 7, 11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Luvisetto, S.; Pietrobon, D.; Azzone, G.F. Uncoupling of oxidative phosphorylation. 1. Protonophoric effects account only partially for uncoupling. Biochemistry 1987, 26, 7332–7338. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, S.C.; Barata, A.G.; Großhans, J.; Teleman, A.A.; Dick, T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011, 14, 819–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, J.R.; Mukherjee, B.; Rogers, B.C.; Nafar, F.; Gosse, M.; Parsons, M.P. The relationship between glutamate dynamics and activity-dependent synaptic plasticity. J. Neurosci. 2020, 40, 2793–2807. [Google Scholar] [CrossRef]
- Penn, A.C.; Zhang, C.L.; Georges, F.; Royer, L.; Breillat, C.; Hosy, E.; Petersen, J.D.; Humeau, Y.; Choquet, D. Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 2017, 549, 384–388. [Google Scholar] [CrossRef]
- Findley, C.A.; Bartke, A.; Hascup, K.N.; Hascup, E.R. Amyloid beta-related alterations to glutamate signaling dynamics during Alzheimer’s disease progression. ASN Neuro 2019, 11, 1759091419855541. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Dysregulation of tau phosphorylation in mouse brain during excitotoxic damage. J. Alzheimer’s Dis. 2009, 17, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Herman, M.A.; Jahr, C.E. Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 2007, 27, 9736–9741. [Google Scholar] [CrossRef]
- Walsh, C.; Barrow, S.; Voronina, S.; Chvanov, M.; Petersen, O.H.; Tepikin, A. Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta 2009, 1787, 1374–1382. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Drago, I.; Filadi, R.; Pozzan, T. Mitochondrial Ca2+ homeostasis: Mechanism, role, and tissue specificities. Pflug. Arch. 2012, 464, 3–17. [Google Scholar] [CrossRef]
- Greotti, E.; Fortunati, I.; Pendin, D.; Ferrante, C.; Galla, L.; Zentilin, L.; Giacca, M.; Kaludercic, N.; Di Sante, M.; Mariotti, L.; et al. mCerulean3-based cameleon sensor to explore mitochondrial Ca2+ dynamics in vivo. iScience 2019, 16, 340–355. [Google Scholar] [CrossRef] [Green Version]
- Petronilli, V.; Cola, C.; Bernardi, P. Modulation of the Mitochondrial Cyclosporin A-Sensitive Permeability Transition Pore. II. The Minimal Requirements for Pore Induction Underscore a Key Role for Transmembrane Electrical Potential, Matrix pH, and Matrix Ca2+—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/7678245/ (accessed on 23 June 2021).
- Szabo, I.; Bernardi, P.; Zoratti, M. Modulation of the mitochondrial megachannel by divalent cations and protons. J. Biol. Chem. 1992, 267, 2940–2946. [Google Scholar] [CrossRef]
- Sweeney, Z.K.; Fu, J.; Wiedmann, B. From chemical tools to clinical medicines: Nonimmunosuppressive cyclophilin inhibitors derived from the cyclosporin and sanglifehrin scaffolds. J. Med. Chem. 2014, 57, 7145–7159. [Google Scholar] [CrossRef] [PubMed]
- Hansson, M.J.; Mattiasson, G.; Månsson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.-M.; Besseghir, K.; Elmér, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, P.; Fovez, Q.; Germain, N.; Khamari, R.; Kluza, J. Mitochondrial spare respiratory capacity: Mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 2020, 34, 13106–13124. [Google Scholar] [CrossRef]
- Maurer, I.; Zierz, S.; Möller, H.-J. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 2000, 21, 455–462. [Google Scholar] [CrossRef]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F 1 F 0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-β and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Emmerzaal, T.L.; Rodenburg, R.J.; Tanila, H.; Verweij, V.; Kiliaan, A.J.; Kozicz, T. Age-dependent decrease of mitochondrial complex II activity in a familial mouse model for Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 66, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Strauss, M.; Hofhaus, G.; Schröder, R.R.; Kühlbrandt, W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 2008, 27, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenaz, G.; Baracca, A.; Barbero, G.; Bergamini, C.; Dalmonte, M.E.; del Sole, M.; Faccioli, M.; Falasca, A.; Fato, R.; Genova, M.L.; et al. Mitochondrial Respiratory Chain Super-Complex I–III in Physiology and Pathology|Elsevier Enhanced Reader. Available online: https://0-reader-elsevier-com.brum.beds.ac.uk/reader/sd/pii/S0005272810000368?token=B01B254C181842A8A9B92B51BC0C5C1BDF3F23D059E5DD736A23C5023209AAD183CD01E52F954B3E848A9ECA700EB856&originRegion=eu-west-1&originCreation=20210626143920 (accessed on 26 June 2021).
- Davies, K.M.; Strauss, M.; Daum, B.; Kief, J.H.; Osiewacz, H.D.; Rycovska, A.; Zickermann, V.; Kuhlbrandt, W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 14121–14126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C. Mitochondrial consumption of cytosolic ATP: Not so fast. FEBS Lett. 2011, 585, 1255–1259. [Google Scholar] [CrossRef] [Green Version]
- Ong, W.-Y.; Tanaka, K.; Dawe, G.S.; Ittner, L.M.; Farooqui, A.A. Slow excitotoxicity in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 35, 643–668. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Veres, G.; Szalárdy, L.; Klivényi, P.; Vécsei, L. Alzheimer’s disease: Recent concepts on the relation of mitochondrial disturbances, excitotoxicity, neuroinflammation, and kynurenines. J. Alzheimer’s Dis. 2018, 62, 523–547. [Google Scholar] [CrossRef] [Green Version]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef]
- Fagian, M.M.; da Silva, L.P.; Vercesi, A.E. Inhibition of oxidative phosphorylation by Ca2+ or Sr2+: A competition with Mg2+ for the formation of adenine nucleotide complexes. Biochim. Biophys. Acta Bioenerg. 1986, 852, 262–268. [Google Scholar] [CrossRef]
- Fink, B.D.; Bai, F.; Yu, L.; Sivitz, W.I. Regulation of ATP production: Dependence on calcium concentration and respiratory state. Am. J. Physiol. Physiol. 2017, 313, C146–C153. [Google Scholar] [CrossRef] [Green Version]
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719. [Google Scholar] [CrossRef]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef]
- Duchen, M.R. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 1992, 283, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Berndt, N.; Kann, O.; Holzhütter, H.-G. Physiology-based kinetic modeling of neuronal energy metabolism unravels the molecular basis of NAD(P)H fluorescence transients. J. Cereb. Blood Flow Metab. 2015, 35, 1494–1506. [Google Scholar] [CrossRef]
- Vergun, O.; Keelan, J.; Khodorov, B.I.; Duchen, M.R. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J. Physiol. 1999, 519, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Filadi, R.; Pizzo, P.; Duchen, M.R. Excitotoxicity revisited: Mitochondria on the verge of a nervous breakdown. Trends Neurosci. 2021, 44, 342–351. [Google Scholar] [CrossRef]
- Boyman, L.; Williams, G.S.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent or Resource | Supplier | Identifier |
|---|---|---|
| Chemicals, peptides, cell culture media and supplements | ||
| Poly-D-Lysine | Merck | P6407 |
| Laminin | Merck | L2020 |
| Minimum Essential Medium, MEM | Gibco | 32360-026 |
| Basal Medium Eagle, BME | Gibco | 41010-026 |
| N2-supplement | Thermo-Fisher | 17502048 |
| B27-supplement | Thermo-Fisher | 17504044 |
| Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) | Merck | C2920 |
| Tetramethylrhodamine, methyl ester (TMRM) | Thermo Fisher | I34361 |
| MitoSOX™ Red | Thermo Fisher | M36008 |
| Cyclosporin H | Merck | SML1575 |
| Bicinchoninic Protein Assay Kit, Quantum Protein | Euroclone | EMP014500 |
| Chemiluminescent reagent Westar Sun | Cyagen | XLS063 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo-Fisher | H1399 |
| MOWIOL® 4-88 Reagent, Poly(vinyl alcohol) | Millipore | 475904-M |
| Antibodies | ||
| Mito profile Total Oxphos rodent WT | Abcam | ab110413 |
| anti HSP 90 | BD Bioscience | 610418 |
| anti-neurofilament | Merck | N5389 |
| anti glial fibrillary acidic protein | Dako | Z0334 |
| Alexa-Fluor Plus 555 goat anti-mouse IgG | Thermo-Fisher | A32727 |
| Alexa Fluor Plus 488 goat anti-rabbit IgG | Thermo-Fisher | A32731 |
| Recombinant proteins | ||
| jRGECO1 | Addgene | 100854 |
| 4mtD3cpv | Addgene | 36324 |
| mito roGFP2-Orp1 | Addgene | 65001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigotto, G.; Zentilin, L.; Pozzan, T.; Basso, E. Effects of Mild Excitotoxic Stimulus on Mitochondria Ca2+ Handling in Hippocampal Cultures of a Mouse Model of Alzheimer’s Disease. Cells 2021, 10, 2046. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10082046
Rigotto G, Zentilin L, Pozzan T, Basso E. Effects of Mild Excitotoxic Stimulus on Mitochondria Ca2+ Handling in Hippocampal Cultures of a Mouse Model of Alzheimer’s Disease. Cells. 2021; 10(8):2046. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10082046
Chicago/Turabian StyleRigotto, Giulia, Lorena Zentilin, Tullio Pozzan, and Emy Basso. 2021. "Effects of Mild Excitotoxic Stimulus on Mitochondria Ca2+ Handling in Hippocampal Cultures of a Mouse Model of Alzheimer’s Disease" Cells 10, no. 8: 2046. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10082046