
The Interplay between Glioblastoma and Its Microenvironment
by
 ,
,
Mark Dapash
1,2 ,
,
David Hou
1,
Brandyn Castro
1,3,
Catalina Lee-Chang
1,4,* and
Maciej S. Lesniak
1,4,* 1
Department of Neurological Surgery, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
2
Pritzker School of Medicine, University of Chicago, Chicago, IL 60637, USA
3
Department of Neurosurgery, University of Chicago, Chicago, IL 60637, USA
4
Northwestern Medicine Malnati Brain Tumor Institute of the Lurie Comprehensive Cancer Center, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(9), 2257; https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092257
Submission received: 30 July 2021
/
Revised: 26 August 2021
/
Accepted: 27 August 2021
/
Published: 31 August 2021
(This article belongs to the Special Issue Immune Microenvironment of Gliomas)
{kind=link}
{kind=link}
Abstract
:GBM is the most common primary brain tumor in adults, and the aggressive nature of this tumor contributes to its extremely poor prognosis. Over the years, the heterogeneous and adaptive nature of GBM has been highlighted as a major contributor to the poor efficacy of many treatments including various immunotherapies. The major challenge lies in understanding and manipulating the complex interplay among the different components within the tumor microenvironment (TME). This interplay varies not only by the type of cells interacting but also by their spatial distribution with the TME. This review highlights the various immune and non-immune components of the tumor microenvironment and their consequences f the efficacy of immunotherapies. Understanding the independent and interdependent aspects of the various sub-populations encapsulated by the immune and non-immune components will allow for more targeted therapies. Meanwhile, understanding how the TME creates and responds to different environmental pressures such as hypoxia may allow for other multimodal approaches in the treatment of GBM. Ultimately, a better understanding of the GBM TME will aid in the development and advancement of more effective treatments and in improving patient outcomes.
1. Introduction
Occurring with an incidence of 3.19 per 100,000 persons, glioblastoma (GBM) represents the most common primary brain tumor in adults [1]. Even with the scientific advances that have allowed for greater knowledge of genomics, molecular biology, and more targeted therapies, patient outcomes remain poor, with a median survival following diagnosis of approximately 14 months [2]. The Stupp Protocol, comprising surgical resection plus adjuvant radiation therapy and temozolomide chemotherapy, followed by temozolomide has remained the standard of care since 2005 [3]. GBM patients with methylation of the O-6-methylguanine-DNA methyltransferase (MGMT) promoter, a DNA repair gene, have shown improved response to temozolomide chemotherapy, leading to improved patient outcomes [4]. The heterogeneity and adaptability of these tumors are the main contributors to their resistance to various therapeutic modalities. In 2010, data from The Cancer Genome Atlas (TCGA) were used to create a novel molecular classification system in which GBM could be group into proneural, classical, or mesenchymal subtypes. In 2010, four molecular subtypes of GBM were described as classical, mesenchymal, neural, and proneural [5]. Key genetic modifications were used to characterize these subtypes including epidermal growth factor receptor (EGFR) mutation/amplification/overexpression in the classical subtype, neurofibromin 1 (NF1) mutations/deletions in the mesenchymal subtype, EGFR amplification/overexpression in the neural subtype, and platelet-derived growth factor receptor alpha (PDGFRa) amplification in the proneural subtype [6]. A majority of GBM arise de novo from a specific trigger mutation in a glioma stem cell (GSC), known as primary GBMs. Secondary GBMs comprise a smaller percentage of GBMs and arise from lower-grade gliomas that acquire additional individual mutations. GBMs that arise from mutations in isocitrate dehydrogenase 1 (IDH1), commonly secondary GBMs, have a better prognosis than GBMs with wild-type IDH1 [7].
In addition to the previously mentioned genetic factors and mutational burden that affects prognosis, the GBM microenvironment contributes significantly to the dynamic and heterogeneous nature of GBM. The tumor microenvironment (TME) describes the active milieu of a tumor and is composed of stromal cells, signaling molecules, immune cells, and the surrounding extracellular matrix (ECM). The heterogeneous nature of GBM and the complex interplay among the different cell populations within its TME have wide-reaching implications (Figure 1). This dynamic interplay contributes to the establishment of hypoxic and necrotic tumor regions, infiltration into the surrounding parenchyma, resistance to radio-chemotherapy, and vascular proliferation [5]. Moreover, despite tumor infiltration via lymphocytes, GBM is considered to be a “cold tumor” due to its high amounts of regulatory B and T cells as well as immunosuppressive myeloid cells. Altogether, this creates a challenging setting for any immunotherapy to be fully efficacious against GBM, and there remains a need for a better understanding of the TME and its contributions towards the clinical picture seen in GBM. This review will summarize interactions within the TME and discuss their implications for the efficacy of immunotherapy.
2. The Extracellular Matrix
In a healthy individual, the extracellular matrix (ECM) is composed of various types of proteins and polysaccharides, which make up roughly 20% of the total volume in an adult human brain [8]. These macromolecules interact with neurons, astrocytes, and other cells to ultimately affect nearly all aspects of development and function. In the brain, various cell types contribute to ECM production, maturation, and structure, whereas ECM proteins in many other tissues are exclusively synthesized and deposited by fibroblasts and other mesenchymal cells [9]. In the setting of malignancies, the changes imposed by the tumor result in the ECM undergoing compositional changes based on their parent cell types. Typically, in the healthy human brain, the ECM is made up of a large proportion of proteoglycans (such as lecticans), glycoproteins (such as tenascin), and glycosaminoglycans (such as hyaluronan) as well as collagen and other fibrous matrix proteins [10]. In the setting of GBM, the composition of the ECM changes, which is physically reflected by the increased stiffness of the tumor-associated ECM [11]. Previous literature has highlighted that increased secretion of ECM components such as hyaluronic acid (HA), fibronectin, thrombospondin, and tenascin-C by glioma cells contribute to this change in ECM composition [12]. The increase of fibronectin and HA in the ECM, as well as the increased expression of particular receptors and integrins on the tumor cell, allow for increased mobility and invasiveness of glioma cells. For example, glioma cells can increase their expression of CD44, the main surface receptor for HA that also binds to matrix metalloproteinase 9 (MMP9) found in the ECM [13]. One specific component of the ECM, mesenchymal stromal cells (MSCs), play an important role in tumor migration. MSCs within the TME are able to release cytokines such as IL-6, CXCL1, and CXCL2, as well as metalloproteinases (MMPs), and contribute to the degradation of the local extracellular matrix [14,15]. Physical changes within the TME such as edema and cellular compression act as physical stressors for cells that cause increased stiffness of the tumor and can contribute to gliomagenesis. Interestingly, murine models have also demonstrated immunomodulation that occurs via the interaction of the innate immune system with the ECM of the tumor. The tumor ECM was shown to be intricately linked to CD47-mediated macrophage phagocytosis signaling through the expression of tumor-associated extracellular matrix protein tenascin C (TNC) [16].
3. The Blood–Brain Barrier
Another important non-immune component of the TME is the blood–brain barrier (BBB). The BBB is a unique attribute of the brain that allows for the tight regulation of molecules and cells [17]. The BBB is formed via the interaction of astrocyte foot processes with endothelial cells and pericytes. The BBB is considered one of the contributors to the poor chemotherapeutic efficacy often seen when treating brain malignancies with intravenous agents [18,19,20]. In the setting of GBM, the BBB is compromised not only because of inflammation and physical distortion but also because of the increased vascularity that contributes to the leakiness of blood vessels [21,22]. The dystrophic growth of the vasculature contributes to the heterogeneity of permeable vessel walls as well as to increased perfusion of the tumors. This increased angiogenesis is primarily due to the high amounts of vascular endothelial growth factor (VEGF) in the TME. Bevacizumab, an antibody that inhibits VEGF, was a therapy that initially showed promise against GBM but ultimately did not improve overall survival. Much of the false hope was due to the anti-angiogenic agent decreasing the leakiness of the vessel walls, leading to less gadolinium contrast within the tumors and the appearance of a smaller tumor on imaging. VEGF, interestingly, also causes the reduction of immune cell extravasation via the reduction in intracellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1) adhesion [23,24]. Ultimately, the BBB becomes compromised and leaky, allowing for the influx of some immune cells. Poor blood flow within the central necrotic areas of the tumors decreases oxygen delivery, contributing to the establishment of hypoxic regions that attract macrophages and promote their immunosuppressive phenotype [25,26].
4. The Central Nervous System Resident Cells
In addition to neurons, the glial compartment in the healthy adult brain comprises astrocytes, oligodendrocytes, and microglia, which serve various functions such as maintenance of the BBB, myelination of axons, and immune surveillance, respectively [27]. In the presence of GBM, the glial compartment within the TME undergoes numerous changes. Although oligodendrocyte-like cells have been found in many pathologic GBM sections, the extent to which these cells contribute to gliomagenesis requires further elucidation. During the growth of the tumor, healthy astrocytes are displaced by GBM-associated astrocytes, further contributing to the weakening of the BBB. Astrocytes within the GBM TME have been shown to undergo reactive astrogliosis similar to phenotypic changes undergone by astrocytes following a traumatic brain injury [28]. This reactive astrogliosis has been noted to contribute to tumor cell infiltration through the activation of zinc finger E-box-binding homeobox 1 (ZEB1), an epithelial–mesenchymal transition (EMT) transcription factor [29,30]. These tumor-associated astrocytes have also been highlighted for their involvement in modulating the immune system within the tumor microenvironment [14]. In fact, through the utilization of programmed death ligand 1 (PD-L1), tumor-associated astrocytes enhance immune suppression within the TME [28]. Microglia also exhibit an intricate interplay with cells encapsulated within the TME. Microglia are recruited to the TME by pro-migratory signals such as granulocyte-macrophage colony-stimulating factor (GM-CSF), stromal derived factor-1 (SDF-1), and glial cell line-derived neurotrophic factor (GDNF) secreted by tumor cells [31]. Microglia are further affected within the TME through their interactions with glioma cells. It was recently demonstrated that glioma cells use extracellular vesicles to interact directly with microglia [32]. This interaction causes genotypic and phenotypic changes that decrease their anti-tumor activity. Similarly, neurons, which also make up the glial compartment, have been shown to promote glioma progression [33,34]. Along with tumor-associated macrophages (TAMs), microglia largely contribute to immunosuppression through the interaction and synergistic release of soluble factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF), C-X3-C Motif Chemokine Ligand 1, (CX3CL1), and SDF1 [35].
5. GBM Cells and Glioma Stem Cells
The communication between GBM cells and the TME is crucial for the proliferation, migration, and immunosuppression of the TME. One key chemokine utilized by tumor cells is C–C motif chemokine ligand 2 (CCL2), which functions to enhance angiogenesis and attract macrophages and microglia to the TME, further contributing to tumor growth [36]. GBM cells also secrete CXCL8, which can function to alter the ECM through the activation of MMPs within the TME [37,38]. GBM cells are also able to interact with microglia and increase their invasiveness by activating TGFβ and EGFR signaling pathways [39]. More recently, extracellular vesicles (EVs) have been highlighted as an important mechanism by which GBM cells communicate bidirectionally with the TME. EVs are used by GBM cells to interact with endothelial cells to promote angiogenesis and with astrocytes to promote ECM degradation [40]. EVs are also used by GBM cells to inhibit apoptosis of astrocytes, further contributing to the aggressiveness of the tumor [41]. Furthermore, EVs incorporating PD-L1 on their surface are able to inhibit T cell activation, further promoting the immunosuppressive environment of the TME [42]. Glioma stem cells (GSCs), like other cancer stem cells, serve as a reservoir for self-renewal and differentiation within the tumor. The differentiation into many unique cell lineages contributes to the heterogeneity seen in GBM and, consequently, to the decreased sensitivity to chemoradiotherapy [43,44,45,46]. In fact, subpopulations of GSCs within the TME have been shown to contribute to the differing susceptibility of GBMs to immunotherapy [47]. GSCs interact with endothelial cells, which results in an enhancement of stemness markers. These cells can be identified via several cell surface markers such as CD133, CD15/SSEA, CD44, and A2B5, although the heterogeneity of these cells prevents one marker from identifying and allow therapeutical targeting of all GSCs [48]. GSCs also contribute to the infiltrative nature of GBM, as studies have shown an association between the number of GSCs in the tumor bulk and the degree of invasiveness [49,50,51]. Their invasive capabilities are in fact enhanced in the presence of TAMs through TGF-β signaling.
6. Immune Cells
6.1. Tumor-Associated Myeloid Cells
A large portion of the tumor mass consists of immune cells. Tumor-associated myeloid cells (TAMCs) can make up as much as 50% of the tumor bulk [52]. TAMCs represent a heterogenous population composed of dendritic cells (DCs), neutrophils, bone marrow-derived macrophages (BMDMs), microglia, and myeloid-derived suppressor cells (MDSCs). It is thought that the majority of these tumor-infiltrating immune cells originate in the periphery rather than from the innate immune cells of the CNS [53]. Within the TAMC cellular compartment, TAMs are one of the most numerous subtypes, consisting of both microglia and bone-marrow-derived macrophages (BMDMs). The quantity of TAMs present within the TME correlates with tumor grade and inversely correlates with overall survival in patients with recurrent GBM [54,55]. TAMs can either release immunosuppressive factors such as interleukin 10 (IL-10) and transforming growth factor beta (TGF-β) or release anti-tumor-promoting factors such as IL-12, TNF-α, depending on the conditions within the TME [56,57]. Using a murine model, recent work has elucidated the phenotypic differences of TAMs based on their origins. TAMs derived from microglia are large, immobile cells with wide arrays of processes extending into the tumor, whereas TAMs derived from monocytes are small and mobile. These unique populations have been demonstrated in human GBM as well [35,58]. MDSCs are another heterogeneous population of cells that impart potent immunosuppressive effects on the TME. MDSCs can be divided into two subtypes: monocytic MDSCs (m-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs). Although similar in their immunosuppressive functions, each subtype has its own unique genomic profile [59]. M-MDSCs contribute to the overall pool of TAMs within the TME and aid in their aggregation within the tumor bulk. M-MDSCs within the hypoxic regions of the tumor undergo phenotypic changes that result in their differentiation into TAMs through the HIF-1α signaling pathway [60]. Similar to TAMs, increased quantities of MDSCs are correlated with the grade of the glioma as well as with a poor prognosis [61,62]. Likewise, at the time of recurrence, high percentages of MDSCs can be used as a poor predictive marker [63]. It has been highlighted that the majority of murine GBM-associated MDSCs in a murine model are M-MDSCs, yet the majority of MDSCs found in patient-derived GBMs are PMN-MDSCs [59]. A keyway in which MDSCs can suppress CD8+ T cell activity is through the increased catabolism of L-arginine. By using arginase-1 and inducible NO synthase, MDSCs utilize L-arginine in the surrounding TME, preventing T cells from utilizing this important amino acid and thus preventing proliferation [64]. They are also able to render the T cell receptor (TCR) non-functional by creating reactive oxygen species (ROS) that cause the nitration of the receptors [65]. Finally, MDSCs also contribute to the pool of IL-10 and TGF-β within the TME, further leading to immunosuppression.
6.2. Tumor-Associated Neutrophils
Neutrophils are another population of TAMCs that is found to accumulate within the GBM TME. Although not as numerous as MDSCs or TAMs, tumor-associated neutrophils (TANs) have been negatively associated with the prognosis of patients with GBM, and the quantity of TANs can serve as a negative prognostic marker for resistance to bevacizumab in patients who did not receive steroids [66]. TANs are typically found in the center of the tumor bulk and are attracted to the TME via macrophage migration inhibitory factor (MIF), C–X–C motif chemokine ligand 8 (CXCL8), and interleukin 8 (IL-8) [67]. TANs aid in tumor progression through their secretion of elastase, which functions to promote tumor proliferation and angiogenesis [68]. To a minor degree, TANs also contribute to the immunosuppressive TME via the secretion of arginase-1, granulocyte colony stimulating factor (G-CSF), and S100 calcium binding protein A4 (S100A4) [66,69].
6.3. Foxp3+ Regulatory T Cells
T cells play a vital role in the adaptive immune response to malignancies. Regulatory T cells (Tregs) are a unique population of T cells that serve to modulate the overall immune homeostasis through immunosuppressive measures. Of particular importance are Tregs that express Forkhead Box P3 (FOXP3) transcription factor. This transcription factor can downregulate the NFAT and NFκB signaling pathways, which consequently downregulates the expression of important effector cytokines such as IL2 [70,71]. A worse prognosis in GBM is associated with a higher Treg-to-T effector cell ratio [72,73]. Tregs are believed to be recruited to the TME via cytokines such as CXCL9/10/11-CXCR3 and CCL5-CCR5 [74]. These cytokines are secreted by innate immune cells within the CNS and glioma cells. Once within the TME, Tregs are subjected to favorable conditions which allow for increased viability and expansion, in addition to promoting the transition of other T cells into Tregs via cytokines such as tumor-derived IL-10 and TGF-β. Tregs themselves also secrete IL-10 and TGF-β [75,76,77] to further promote immunosuppression. These molecules are able to exert an immunosuppressive effect on natural killer (NK) cells, aid in the generation of MDSCs, and impair the antigen-presenting ability of DCs. Tregs also highly express key immune checkpoint molecules such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed-death 1 (PD-1), and glucocorticoid-induced TNFR family-related gene (GITR) [78,79,80]. These molecules interact with their respective receptors on the surface of other immune cells to suppress the cells’ effector activities. Thus far, clinical trials investigating immune checkpoint inhibitors have mainly focused on targeting CTLA-4 and PD-1 in order modulate the anti-tumor response [81,82,83]. The use of T cells as immunotherapeutic tools has been explored in various malignancies including GBM. Chimeric antigen receptor (CAR) T cell therapy has been heavily investigated in several types of cancers [84,85,86,87]. These engineered T cells utilize tumor-associated antigens (TAAs) to allow T cells to become activated and gain greater specificity against tumor cells. TAAs such as variant III of the EGFR (EGFRvIII), human epidermal growth factor receptor 2 (HER2), or interleukin 13 (IL-13) receptor α2 (IL-13Rα2) have been investigated as possible targets [88,89,90,91]. More recent studies have investigated the efficacy of bispecific and trivalent CAR T cells [92]. Trivalent CAR T cells targeting HER2, IL13Rα2, and ephrin-A2 (EphA2) demonstrated improved cytotoxicity when compared to monospecific or bispecific CAR T cells [93]. Nevertheless, these T cells are subjected to the harsh TME of GBM and require further development.
6.4. Natural Killers
The innate immune system also contributes to the unique nature of the tumor microenvironment. NK cells are an important part of the innate immune system and are critical for the antitumor immune response, particularly through their interactions with major histocompatibility complex class I molecules (MHC-I). NK cells use granzyme B and perforin to provoke cellular apoptosis through contact-dependent cytotoxicity [94]. NK cells have been identified as part of the population of immune cells that infiltrate the GBM TME [95]. It has been shown that GBM with the R132H mutation in IDH1 contains neurons that can recruit NK cells to the CNS via the CX3CL1 chemokine [96]. NK cells have also been shown to be able to control tumor growth through cytokine secretion which is promoted via the NKp44 receptor. PDGF-D is expressed by most GBMs and binds to the activating NKp44 receptor to stimulate cytokine secretion from NK cells and innate lymphoid cells [97]. Tumor progression is also associated with B7-H6, which is known to augment NK cell functionality through the activation of their NKp30 receptors [98]. Nevertheless, NK cells are subjected to immunosuppressive factors within the TME. One key way by which the antitumor functionality of NK cells is suppressed is through cellular contact with glioma cells. Glioma cells can express unique MHC-I molecules that can bind to receptors on the surface of NK cells, thus suppressing their functions [99]. NK cells within the TME have been shown to regulate the levels of IFN-γ, which in turn can promote GSCs differentiation [100]. Furthermore, this change allows GSCs to become more resistant to NK cell cytotoxicity [101]. It has also been observed that radio-chemotherapy decreases the quantity of tumor-infiltrating NK cells [102]. The therapeutic potential of NK cells has been investigated by creating cytokine-induced killer (CIK) cells. CIK cells are created via culturing NK cells with IFN-γ, IL-2, and anti-CD3 monoclonal antibody (CD3 mAb) [103]. When tested in an open-labeled phase III clinical trial based in South Korea, this treatment did not show a significant difference in patient’s overall survival, highlighting the need for further investigation [104].
6.5. Dendritic Cells
Dendritic cells (DCs) are a class of antigen-presenting cells that serve as a vital bridge between the innate and the adaptive immune systems. DCs are important in monitoring pathogens or inflammatory responses throughout the body. DCs can endocytose, process, and present antigens to B and T cells, promoting their activation [105]. DCs are typically found in the meninges and the choroid plexus, but not within the healthy brain parenchyma [106]. In the setting of chronic inflammation, like that seen in GBM, DCs have been found within the brain. It has been shown in murine models that DCs are recruited to the TME via chemokines such as CCL5 and XCL1, similar to NK cells [107]. NK cells are also able to recruit DCs to the TME through the use of CCL5 and XCLI [107]. DCs have been shown to produce antitumor-augmenting cytokines such as IL12, which in turn can recruit more CD8+ T cells and reinvigorate anergic T cells [108]. Nevertheless, DCs are subjected to immunosuppressive effects from the TME which can induce a regulatory phenotype. These regulatory DCs can in turn activate Tregs and downregulate the recruitment of CD8+ T cells [109]. DCs have been a topic of interest for the development of new vaccines against GBM. Previous studies have highlighted the efficacy of DC-based vaccines in preclinical models as well as early-stage clinical trials [110,111,112]. Still, no successful phase III trials have been completed utilizing this type of vaccine.
6.6. B Cells
The role of B cells in antitumor immunity has remained nebulous in human and in in vivo studies. For one, tumor-infiltrating B cells have been correlated with poor prognosis of patients with metastatic carcinomas, while patients with breast carcinoma had improved survival [113,114]. Still, B cells are critical in their role as antigen-presenting cells and for their ability to induce clonal expansion of T cells sensitized to TAAs. B cells expressing 4-1BBL+ costimulatory surface protein have also been shown to be able to improve the functionality and viability of CD8+ T cells [115]. They do this by the secretion of cytokines such as TNFα [116]. Research efforts have now highlighted that regulatory B cells can also be found within the TME. Regulatory B cells were identified to overexpress immunosuppressive molecules such as PD-L1 and CD155 in addition to producing IL-10 and TGF-β [117] as a result of microvesicle-mediated interactions with MDSC. MDSCs were shown to secrete microvesicles containing PD-L1, which is then endocytosed by tumor-infiltrating B cells [52]. These B cells then act as potent inhibitors of CD8+ T cell cytotoxicity.
7. Implications of the TME on Immunotherapies
Immune checkpoint inhibitors (ICIs) are monoclonal antibodies which target inhibitory ligands and their receptors which are commonly expressed by GBM cells, lymphocytes, and myeloid cells [118]. Consequently, ICIs allow for a more robust antitumor response by CD8+ T cells. Most research into ICIs has focused on PD-1, PD-L1, and CTLA-4, important components of immune checkpoints pathways [78,80]. The use of ICIs has led to increased survival when compared to chemotherapy in patients with various tumors; however, it has had limited efficacy in the treatment of GBM in clinical trials [119,120,121]. Tumor vaccines utilize one or several tumor-associated antigens (TAAs) to stimulate a cellular and/or humoral immune response. Dendritic-based vaccines are created utilizing dendritic cells primed with whole cell tumor lysates or particular TAAs, while peptide-based vaccines activate T cells through the use of short amino acids [122]. For example, the variant III of the epidermal growth factor receptor (EGFRvIII) is a commonly acknowledged target for the use of peptide-based vaccination in GBM [123]. Clinical trials on both peptide- and dendritic-based vaccines have shown limited clinical benefits, despite the ability of these two strategies to invoke immune responses [112,124,125,126]. Another immunotherapeutic modality employs T cells with modified tumor-specific T cell receptors (TCRs) or chimeric antigen receptors (CARs) to generate a more precise anti-tumor response [127]. Currently, early-stage clinical trials involving CAR T cell therapy have shown safety and encouraging immune responses [128]. The various components of the GBM TME contribute to the limited efficacy of the current immunotherapeutic modalities. The non-immune components serve critical roles in the limited efficacy and eventual resistance through the creation of niches within the TME. The BBB and the irregular neovasculature prevent the optimal delivery of drugs, such as ICIs [129]. Furthermore, the inelastic and dense ECM increases metabolic stress and hypoxia, which ultimately activate pathways that inhibit apoptosis in cells closely interacting with the ECM [130]. GSCs are typically found within perivascular niches and closely interact with the ECM to modulate their function [131]. GCS contribute to tumor growth, invasion, and immunosuppression within the TME. The immunosuppressive nature of the TME remains one of the major limitations to achieving an effective anti-tumor response. Components such as regulatory B cells, regulatory T cells, and TAMCs secrete soluble factors such as IL-10 and TGF-β. This further promotes the recruitment and differentiation of additional regulatory B and T cells [132,133]. GBM cells are also able to interact with the TME, particularly the innate immune system, through the use of EVs containing PD-L1 on their surface [134]. These niches and complex interplay among the components of the TME create the intra- and inter-tumor heterogeneity which further limits the efficacy of immunotherapies.
8. Conclusions
The limited efficacy of the current treatments and the associated poor long-term prognosis of GBM is in a large part a consequence of the TME. This review highlights the intricate interplay among the immune and non-immune components of the TME. This interplay contributes to the establishment of a heterogeneous and adaptive TME which ultimately serves to increase the degree of immunosuppression, invasiveness, proliferation in these tumors. The non-immune components, particularly the BBB, as well as neurons, microglia, and ECM are important contributors to the alterations that take place within the TME. Still, the immune component made up of macrophages, DCs, B cells, and T cells can account for the majority of the extensive tumor-promoting effects seen within and outside of the TME. Several therapeutic approaches particularly targeting the TME have led to improved outcomes; however, this has been limited to very select sub-groups of patients. Immunotherapies such peptide-based and cell-based vaccines as well as immune checkpoint inhibitors aim to bolster the adaptive immune system to promote more robust anti-tumor responses. Yet, low tumor immunogenicity and immunosuppressive stressors, as a consequence of the interplay of various components of the TME, ultimately lead to resistance to immunotherapies. More recent work has allowed for further classification and identification of unique subpopulations of cells within the TME, further highlighting its heterogeneous nature. Continued investigation into the TME will aid in our understanding of how these elements contribute to the therapeutic response and interact with one another. This will lead to the creation of a more multimodal, yet targeted approach to the treatment of GBM.
Funding
This research was funded by the National Institutes of Health (NIH), grant number R35CA197725 (M.S.L.), R01NS093903 (M.S.L.) and 1R37CA258426-01 (CLC), the Northwestern SPORE for Translational Approaches to Brain Cancer, grant number P50CA221747 (M.S.L.), the Malnati’s Brain Tumor Institute (CLC), and by the Neurosurgery Research and Education Foundation (BC).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved Survival Time Trends for Glioblastoma Using the SEER 17 Population-Based Registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- DeCordova, S.; Shastri, A.; Tsolaki, A.G.; Yasmin, H.; Klein, L.; Singh, S.K.; Kishore, U. Molecular Heterogeneity and Immunosuppressive Microenvironment in Glioblastoma. Front. Immunol. 2020, 11, 1402. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Lam, D.; Enright, H.A.; Cadena, J.; Peters, S.K.G.; Sales, A.P.; Osburn, J.J.; Soscia, D.A.; Kulp, K.S.; Wheeler, E.K.; Fischer, N.O. Tissue-Specific Extracellular Matrix Accelerates the Formation of Neural Networks and Communities in a Neuron-Glia Co-Culture on a Multi-Electrode Array. Sci. Rep. 2019, 9, 4159. [Google Scholar] [CrossRef]
- Barros, C.S.; Franco, S.J.; Müller, U. Extracellular Matrix: Functions in the Nervous System. Cold Spring Harb. Perspect. Biol. 2011, 3, a005108. [Google Scholar] [CrossRef] [Green Version]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular Matrix Assembly: A Multiscale Deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Wang, C.; Sinha, S.; Jiang, X.; Murphy, L.; Fitch, S.; Wilson, C.; Grant, G.; Yang, F. Matrix Stiffness Modulates Patient-Derived Glioblastoma Cell Fates in Three-Dimensional Hydrogels. Tissue Eng. Part A 2020, 27, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Belousov, A.; Titov, S.; Shved, N.; Garbuz, M.; Malykin, G.; Gulaia, V.; Kagansky, A.; Kumeiko, V. The Extracellular Matrix and Biocompatible Materials in Glioblastoma Treatment. Front. Bioeng. Biotechnol. 2019, 7, 341. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kumar, S. CD44-Mediated Adhesion to Hyaluronic Acid Contributes to Mechanosensing and Invasive Motility. Mol. Cancer Res. 2014, 12, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.-G.; Joyce, C.; et al. Mesenchymal Stem Cells Isolated from Human Gliomas Increase Proliferation and Maintain Stemness of Glioma Stem Cells Through the IL-6/Gp130/STAT3 Pathway. Stem. Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, D.; Giacomelli, C.; Marchetti, L.; Martini, C.; Trincavelli, M.L. High Adenosine Extracellular Levels Induce Glioblastoma Aggressive Traits Modulating the Mesenchymal Stromal Cell Secretome. Int. J. Mol. Sci. 2020, 21, 7706. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, S.; Lal, B.; Wei, S.; Wang, S.; Zhan, D.; Zhang, H.; Lee, R.S.; Gao, P.; Lopez-Bertoni, H.; et al. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, C.; Wang, L.; Chen, Y. A Comprehensive Review in Improving Delivery of Small-Molecule Chemotherapeutic Agents Overcoming the Blood-Brain/Brain Tumor Barriers for Glioblastoma Treatment. Drug Deliv. 2019, 26, 551–565. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood Brain Barrier: A Challenge for Effectual Therapy of Brain Tumors. BioMed Res. Int. 2015, 2015, e320941. [Google Scholar] [CrossRef] [Green Version]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The Disturbed Blood-Brain Barrier in Human Glioblastoma. Mol Asp. Med. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.L. de S. e; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the Vascular Fragility of the Blood Brain Barrier. Front. Cell Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momeny, M.; Moghaddaskho, F.; Gortany, N.K.; Yousefi, H.; Sabourinejad, Z.; Zarrinrad, G.; Mirshahvaladi, S.; Eyvani, H.; Barghi, F.; Ahmadinia, L.; et al. Blockade of Vascular Endothelial Growth Factor Receptors by Tivozanib Has Potential Anti-Tumour Effects on Human Glioblastoma Cells. Sci. Rep. 2017, 7, 44075. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia Promotes Glioma-Associated Macrophage Infiltration via Periostin and Subsequent M2 Polarization by Upregulating TGF-Beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and Macrophages Promote Glioblastoma Invasion by the CCL4-CCR5 Axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-Associated Reactive Astrocytes Aid the Evolution of Immunosuppressive Environment in Glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [Green Version]
- Iser, I.C.; Lenz, G.; Wink, M.R. EMT-like Process in Glioblastomas and Reactive Astrocytes. Neurochem. Int. 2019, 122, 139–143. [Google Scholar] [CrossRef]
- Niklasson, M.; Bergström, T.; Jarvius, M.; Sundström, A.; Nyberg, F.; Haglund, C.; Larsson, R.; Westermark, B.; Segerman, B.; Segerman, A. Mesenchymal Transition and Increased Therapy Resistance of Glioblastoma Cells Is Related to Astrocyte Reactivity. J. Pathol. 2019, 249, 295–307. [Google Scholar] [CrossRef]
- Al-kharboosh, R.; ReFaey, K.; Lara-Velazquez, M.; Grewal, S.S.; Imitola, J.; Quiñones-Hinojosa, A. Inflammatory Mediators in Glioma Microenvironment Play a Dual Role in Gliomagenesis and Mesenchymal Stem Cell Homing: Implication for Cellular Therapy. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 4, 443–459. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Abels, E.R.; Van De Haar, L.L.; Zhang, X.; Morsett, L.; Sil, S.; Guedes, J.; Sen, P.; Prabhakar, S.; Hickman, S.E.; et al. Glioblastoma Hijacks Microglial Gene Expression to Support Tumor Growth. J. Neuroinflamm. 2020, 17, 120. [Google Scholar] [CrossRef]
- Gillespie, S.; Monje, M. An Active Role for Neurons in Glioma Progression: Making Sense of Scherer’s Structures. Neuro Oncol. 2018, 20, 1292–1299. [Google Scholar] [CrossRef]
- Johung, T.; Monje, M. Neuronal Activity in the Glioma Microenvironment. Curr. Opin. Neurobiol. 2017, 47, 156–161. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groblewska, M.; Litman-Zawadzka, A.; Mroczko, B. The Role of Selected Chemokines and Their Receptors in the Development of Gliomas. Int. J. Mol. Sci. 2020, 21, 3704. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mou, L.; Pan, Y.; Feng, C.; Zhang, J.; Li, J. CXCL8 Promotes Glioma Progression By Activating The JAK/STAT1/HIF-1α/Snail Signaling Axis. Onco Targets Ther. 2019, 12, 8125–8138. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Graeber, M.B. The Molecular Profile of Microglia under the Influence of Glioma. Neuro Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the Glioblastoma Micro-Environment via Extracellular Vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma Multiforme-Derived Extracellular Vesicles Drive Normal Astrocytes towards a Tumour-Enhancing Phenotype. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef] [Green Version]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune Evasion Mediated by PD-L1 on Glioblastoma-Derived Extracellular Vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy Resistance of Glioblastoma Stem Cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [Green Version]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therapy. Stem Cells Int. 2016, 2016, e7849890. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The Role of Glioma Stem Cells in Chemotherapy Resistance and Glioblastoma Multiforme Recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.-M. Invasive Glioblastoma Cells Acquire Stemness and Increased Akt Activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated Invasive Potential of Glioblastoma Stem Cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bien-Möller, S.; Balz, E.; Herzog, S.; Plantera, L.; Vogelgesang, S.; Weitmann, K.; Seifert, C.; Fink, M.A.; Marx, S.; Bialke, A.; et al. Association of Glioblastoma Multiforme Stem Cell Characteristics, Differentiation, and Microglia Marker Genes with Patient Survival. Stem Cells Int. 2018, 2018, e9628289. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic Targeting of Tumor-Associated Myeloid Cells Synergizes with Radiation Therapy for Glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef]
- Lu-Emerson, C.; Snuderl, M.; Kirkpatrick, N.D.; Goveia, J.; Davidson, C.; Huang, Y.; Riedemann, L.; Taylor, J.; Ivy, P.; Duda, D.G.; et al. Increase in Tumor-Associated Macrophages after Antiangiogenic Therapy Is Associated with Poor Survival among Patients with Recurrent Glioblastoma. Neuro Oncol. 2013, 15, 1079–1087. [Google Scholar] [CrossRef]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical Debulking Promotes Recruitment of Macrophages and Triggers Glioblastoma Phagocytosis in Combination with CD47 Blocking Immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef] [Green Version]
- Akins, E.A.; Aghi, M.K.; Kumar, S. Incorporating Tumor-Associated Macrophages into Engineered Models of Glioma. iScience 2020, 23, 101770. [Google Scholar] [CrossRef] [PubMed]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; ISBN 978-0-9944381-2-6. [Google Scholar]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of Myeloid-Derived Suppressor Cells in Cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.J.F.H.; ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated Levels of Polymorphonuclear Myeloid-Derived Suppressor Cells in Patients with Glioblastoma Highly Express S100A8/9 and Arginase and Suppress T Cell Function. Neuro Oncol. 2016, 18, 1253–1264. [Google Scholar] [CrossRef]
- Richard, S.A. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis. Markers 2020, 2020, e8844313. [Google Scholar] [CrossRef]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global Immune Fingerprinting in Glioblastoma Patient Peripheral Blood Reveals Immune-Suppression Signatures Associated with Prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raber, P.; Ochoa, A.C.; Rodríguez, P.C. Metabolism of L-Arginine by Myeloid-Derived Suppressor Cells in Cancer: Mechanisms of T Cell Suppression and Therapeutic Perspectives. Immunol. Investig. 2012, 41, 614–634. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression Mediated by Myeloid-Derived Suppressor Cells (MDSCs) during Tumour Progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massara, M.; Persico, P.; Bonavita, O.; Mollica Poeta, V.; Locati, M.; Simonelli, M.; Bonecchi, R. Neutrophils in Gliomas. Front. Immunol. 2017, 8, 1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, X.H.-F. Tumor-Associated Neutrophils and Macrophages—Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef]
- Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 1954. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H. FOXP3 and Its Role in the Immune System. Adv. Exp. Med. Biol. 2009, 665, 17–29. [Google Scholar] [CrossRef]
- Lu, L.; Barbi, J.; Pan, F. The Regulation of Immune Tolerance by FOXP3. Nat. Rev. Immunol. 2017, 17, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and Prognostic Impact of FoxP3+ Regulatory T Cells in Human Gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased Proportion of FoxP3+ Regulatory T Cells in Tumor Infiltrating Lymphocytes Is Associated with Tumor Recurrence and Reduced Survival in Patients with Glioblastoma. Cancer Immunol. Immunother. 2015, 64, 419–427. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Richmond, A. Chemokines Modulate Immune Surveillance in Tumorigenesis, Metastasis, and Response to Immunotherapy. Front. Immunol. 2019, 10, 333. [Google Scholar] [CrossRef] [Green Version]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of Antitumor Immunity by IL-10 and TGF-β-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4+ and CD8+ Regulatory T Cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Humphries, W.; Wei, J.; Sampson, J.H.; Heimberger, A.B. The Role of Tregs in Glioma-Mediated Immunosuppression: Potential Target for Intervention. Neurosurg. Clin. N. Am. 2010, 21, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T Cells in Tumor Microenvironment: New Mechanisms, Potential Therapeutic Strategies and Future Prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Kelly, W.J.; Giles, A.J.; Gilbert, M. T Lymphocyte-Targeted Immune Checkpoint Modulation in Glioma. J. Immunother. Cancer 2020, 8, 379. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Rashidi, A.; Chang, A.L.; Muroski, M.E.; Han, Y.; Zhang, L.; Lesniak, M.S. Anti-GITR Therapy Promotes Immunity against Malignant Glioma in a Murine Model. Cancer Immunol. Immunother. 2016, 65, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role of PD-1/PD-L1 Axis in Treg Development and Function: Implications for Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent Glioma Clinical Trial, CheckMate-143: The Game Is Not over Yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; de Andrea, C.; López-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Tremelimumab and Durvalumab in Combination or Alone in Treating Patients with Recurrent Malignant Glioma—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02794883 (accessed on 22 April 2020).
- Zeltsman, M.; Dozier, J.; McGee, E.; Ngai, D.; Adusumilli, P.S. CAR T-Cell Therapy for Lung Cancer and Malignant Pleural Mesothelioma. Transl. Res. 2017, 187, 1–10. [Google Scholar] [CrossRef]
- Forsberg, E.M.V.; Lindberg, M.F.; Jespersen, H.; Alsén, S.; Bagge, R.O.; Donia, M.; Svane, I.M.; Nilsson, O.; Ny, L.; Nilsson, L.M.; et al. HER2 CAR-T Cells Eradicate Uveal Melanoma and T-Cell Therapy–Resistant Human Melanoma in IL2 Transgenic NOD/SCID IL2 Receptor Knockout Mice. Cancer Res. 2019, 79, 899–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Charging the T Cells to Fight. J. Transl. Med. 2020, 18, 428. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T Cells for Brain Tumors: Lessons Learned and Road Ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational Development and Characterization of Humanized Anti-EGFR Variant III Chimeric Antigen Receptor T Cells for Glioblastoma. Sci. Transl. Med. 2015, 7, 275ra22. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Thaci, B.; Crawford, A.C.; Sampath, P. Interleukin-13 Receptor Alpha 2-Targeted Glioblastoma Immunotherapy. Biomed. Res. Int. 2014, 2014, 952128. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T Cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro-Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Han, S.J.; Sughrue, M.E.; Tihan, T.; Parsa, A.T. Immune Cell Infiltrate Differences in Pilocytic Astrocytoma and Glioblastoma: Evidence of Distinct Immunological Microenvironments That Reflect Tumor Biology. J. Neurosurg. 2011, 115, 505–511. [Google Scholar] [CrossRef]
- Ren, F.; Zhao, Q.; Huang, L.; Zheng, Y.; Li, L.; He, Q.; Zhang, C.; Li, F.; Maimela, N.R.; Sun, Z.; et al. The R132H Mutation in IDH1 Promotes the Recruitment of NK Cells through CX3CL1/CX3CR1 Chemotaxis and Is Correlated with a Better Prognosis in Gliomas. Immunol. Cell Biol. 2019, 97, 457–469. [Google Scholar] [CrossRef]
- Sedgwick, A.J.; Ghazanfari, N.; Constantinescu, P.; Mantamadiotis, T.; Barrow, A.D. The Role of NK Cells and Innate Lymphoid Cells in Brain Cancer. Front. Immunol. 2020, 11, 1549. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wu, W.; Zhang, H.; Zhang, X.; Zhang, D.; Wang, Q.; Huang, L.; Wang, Y.; Hang, C. High Expression of B7-H6 in Human Glioma Tissues Promotes Tumor Progression. Oncotarget 2017, 8, 37435–37447. [Google Scholar] [CrossRef] [PubMed]
- Burster, T.; Gärtner, F.; Bulach, C.; Zhanapiya, A.; Gihring, A.; Knippschild, U. Regulation of MHC I Molecules in Glioblastoma Cells and the Sensitizing of NK Cells. Pharmaceuticals 2021, 14, 236. [Google Scholar] [CrossRef]
- Martin-Hijano, L.; Sainz, B. The Interactions between Cancer Stem Cells and the Innate Interferon Signaling Pathway. Front. Immunol. 2020, 11, 526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowska, A.K.; Tseng, H.-C.; Kaur, K.; Topchyan, P.; Inagaki, A.; Bui, V.T.; Kasahara, N.; Cacalano, N.; Jewett, A. Resistance to Cytotoxicity and Sustained Release of Interleukin-6 and Interleukin-8 in the Presence of Decreased Interferon-γ after Differentiation of Glioblastoma by Human Natural Killer Cells. Cancer Immunol. Immunother. 2016, 65, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Fares, J.; Fares, M.Y.; Fares, Y. Natural Killer Cells in the Brain Tumor Microenvironment: Defining a New Era in Neuro-Oncology. Surg. Neurol. Int. 2019, 10, 43. [Google Scholar] [CrossRef]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID Mouse/Human Lymphoma Model to Evaluate Cytokine-Induced Killer Cells with Potent Antitumor Cell Activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, D.-S.; Nam, D.-H.; Kang, S.-H.; Lee, J.W.; Chang, J.-H.; Kim, J.-H.; Lim, Y.-J.; Koh, Y.-C.; Chung, Y.-G.; Kim, J.-M.; et al. Phase III Randomized Trial of Autologous Cytokine-Induced Killer Cell Immunotherapy for Newly Diagnosed Glioblastoma in Korea. Oncotarget 2016, 8, 7003–7013. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain Dendritic Cells: Biology and Pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.J.; Ornelles, D.A.; Mitchell, L.M.; Brzoza-Lewis, K.L.; Hiltbold, E.M. IL-12 Produced by Dendritic Cells Augments CD8+ T Cell Activation through the Production of the Chemokines CCL1 and CCL17. J. Immunol. 2008, 181, 8576–8584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [Green Version]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.-W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic Cell Vaccination in Glioblastoma Patients Induces Systemic and Intracranial T-Cell Responses Modulated by the Local Central Nervous System Tumor Microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First Results on Survival from a Large Phase 3 Clinical Trial of an Autologous Dendritic Cell Vaccine in Newly Diagnosed Glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical Evaluation of Dendritic Cell Vaccination for Patients with Recurrent Glioma: Results of a Clinical Phase I/II Trial. Clin. Cancer Res. 2005, 11, 4160–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, G.J.; Demissie, E.; Pillai, S. B Lymphocytes and Cancer: A Love-Hate Relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Chang, C.; Bodogai, M.; Moritoh, K.; Olkhanud, P.B.; Chan, A.C.; Croft, M.; Mattison, J.A.; Holst, P.J.; Gress, R.E.; Ferrucci, L.; et al. Accumulation of 4-1BBL+ B Cells in the Elderly Induces the Generation of Granzyme-B+ CD8+ T Cells with Potential Antitumor Activity. Blood 2014, 124, 1450–1459. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B Cells with CD40 Agonism and IFNγ Elicits Potent Immunity against Glioblastoma. J. Exp. Med. 2020, 218, e20200913. [Google Scholar] [CrossRef] [PubMed]
- Lee-Chang, C.; Rashidi, A.; Miska, J.; Zhang, P.; Pituch, K.C.; Hou, D.; Xiao, T.; Fischietti, M.; Kang, S.J.; Appin, C.L.; et al. Myeloid-Derived Suppressive Cells Promote B Cell–Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol. Res. 2019, 7, 1928–1943. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, L.; Ye, M.; Kang, C.; You, H. Treatment Progress of Immune Checkpoint Blockade Therapy for Glioblastoma. Front. Immunol. 2020, 11, 3080. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.; Shaw, H.; Cohn-Brown, D.; Chester, K.; Mulholland, P. Ipilimumab and Bevacizumab in Glioblastoma. Clin. Oncol. 2016, 28, 622–626. [Google Scholar] [CrossRef] [Green Version]
- Kong, Z.; Wang, Y.; Ma, W. Vaccination in the Immunotherapy of Glioblastoma. Hum. Vaccines Immunother. 2017, 14, 255–268. [Google Scholar] [CrossRef]
- Choi, B.D.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; McLendon, R.E.; Bigner, D.D.; Sampson, J.H. EGFRvIII-Targeted Vaccination Therapy of Malignant Glioma. Brain Pathol. 2009, 19, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic Escape after Prolonged Progression-Free Survival with Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients with Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune Response in Patients With Newly Diagnosed Glioblastoma Multiforme Treated With Intranodal Autologous Tumor Lysate-Dendritic Cell Vaccination After Radiation Chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A Phase II, Multicenter Trial of Rindopepimut (CDX-110) in Newly Diagnosed Glioblastoma: The ACT III Study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, L.; Brown, C.; Badie, B. Chimeric Antigen Receptor T-Cell Therapy: Updates in Glioblastoma Treatment. Neurosurgery 2021, 88, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, X.; Qian, Y.; Yuan, X.; Ding, Y.; Hu, D.; He, X.; Wu, Y. Chimeric Antigen Receptor T-Cell Therapy in Glioblastoma: Current and Future. Front. Immunol. 2020, 11, 2837. [Google Scholar] [CrossRef]
- Sanders, S.; Debinski, W. Challenges to Successful Implementation of the Immune Checkpoint Inhibitors for Treatment of Glioblastoma. Int. J. Mol. Sci. 2020, 21, 2759. [Google Scholar] [CrossRef] [PubMed]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Hira, V.V.V.; Aderetti, D.A.; van Noorden, C.J.F. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magaña-Maldonado, R.; Chávez-Cortez, E.G.; Olascoaga-Arellano, N.K.; López-Mejía, M.; Maldonado-Leal, F.M.; Sotelo, J.; Pineda, B. Immunological Evasion in Glioblastoma. BioMed Res. Int. 2016, 2016, e7487313. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.F.; Cui, J.W. The Role of Tumor-Infiltrating B Cells in Tumor Immunity. J. Oncol. 2019, 2019, 2592419. [Google Scholar] [CrossRef] [PubMed]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2019, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
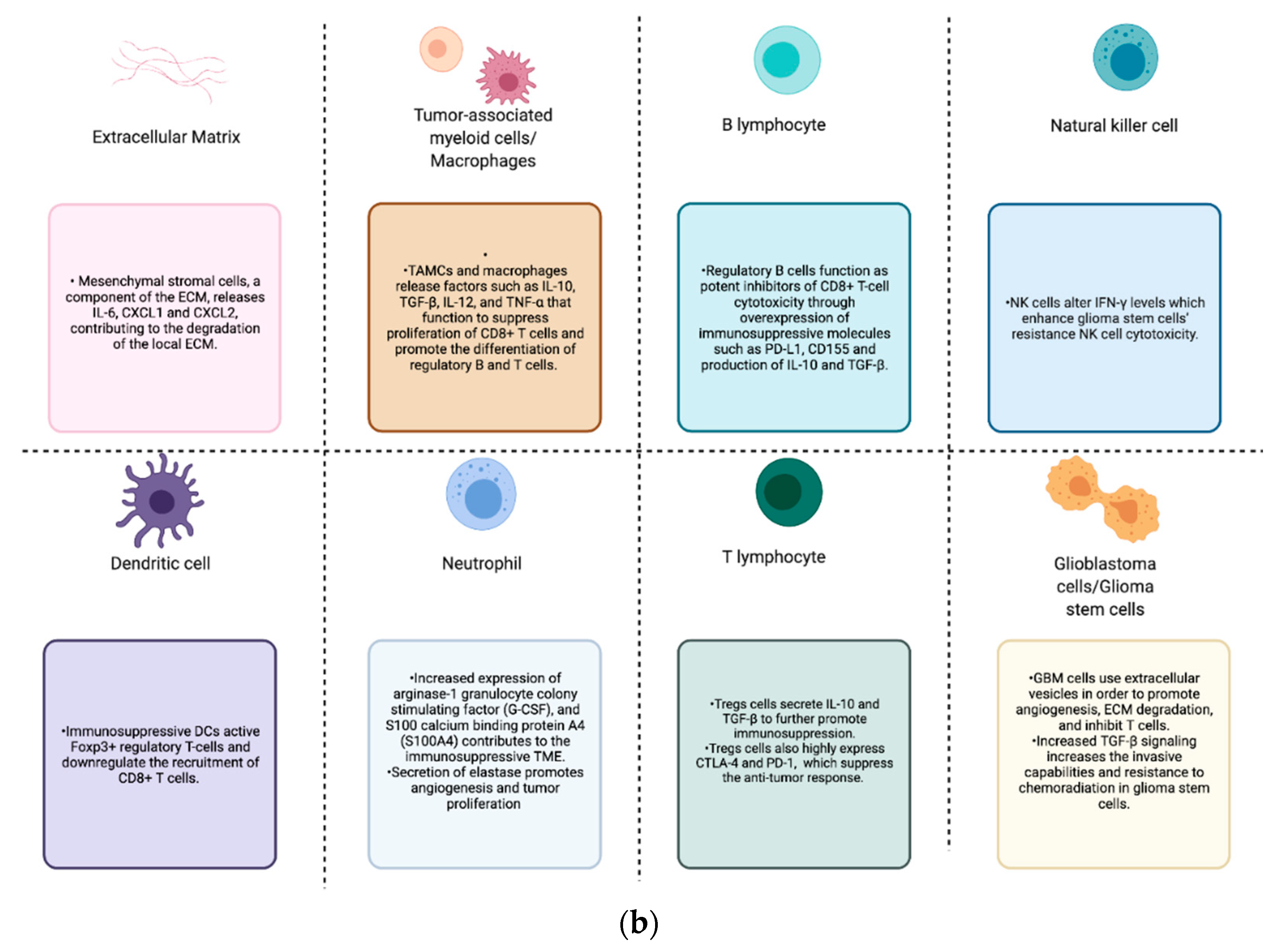
Figure 1.
(a) GBM tumor microenvironment and its components; (b) components of the GBM TME and their contributions to the immunosuppression, proliferation, invasion, and treatment resistance seen in these tumors.
Figure 1.
(a) GBM tumor microenvironment and its components; (b) components of the GBM TME and their contributions to the immunosuppression, proliferation, invasion, and treatment resistance seen in these tumors.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dapash, M.; Hou, D.; Castro, B.; Lee-Chang, C.; Lesniak, M.S. The Interplay between Glioblastoma and Its Microenvironment. Cells 2021, 10, 2257. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092257
AMA Style
Dapash M, Hou D, Castro B, Lee-Chang C, Lesniak MS. The Interplay between Glioblastoma and Its Microenvironment. Cells. 2021; 10(9):2257. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092257
Chicago/Turabian StyleDapash, Mark, David Hou, Brandyn Castro, Catalina Lee-Chang, and Maciej S. Lesniak. 2021. "The Interplay between Glioblastoma and Its Microenvironment" Cells 10, no. 9: 2257. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092257
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.