
Effects of Fasting and Feeding on Transcriptional and Posttranscriptional Regulation of Insulin-Degrading Enzyme in Mice

 ,
, 
 , ,
, ,  , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mouse Studies
2.2. Plasma Biochemistry
2.3. Quantitative Real-Time PCR
2.4. Western Blot Analyses
2.5. IDE Activity
2.6. Statistical Analyses
3. Results
3.1. Metabolic Responses to the Fasting-to-Refeeding Transition in Mice Fed an SD or HFD
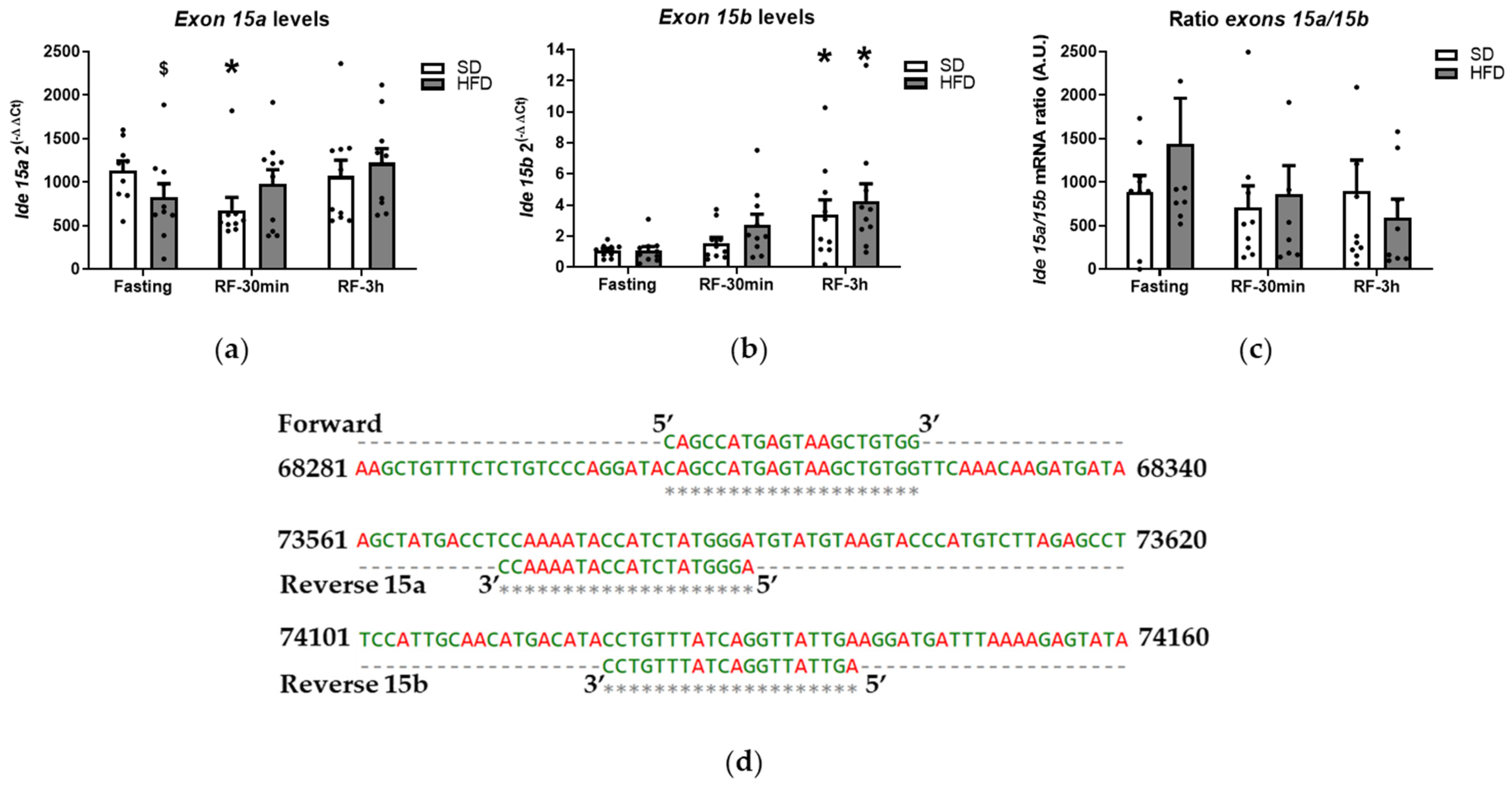
3.2. Liver IDE Expression and Activity
3.3. Kidney IDE Expression and Activity
3.4. Skeletal Muscle IDE Expression and Activity
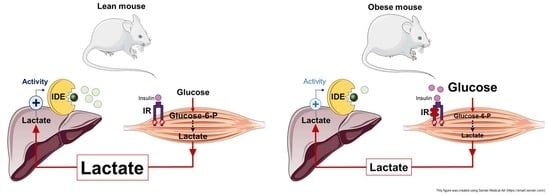
3.5. Reduced Liver and Muscle IDE Levels Associate with Insulin Resistance
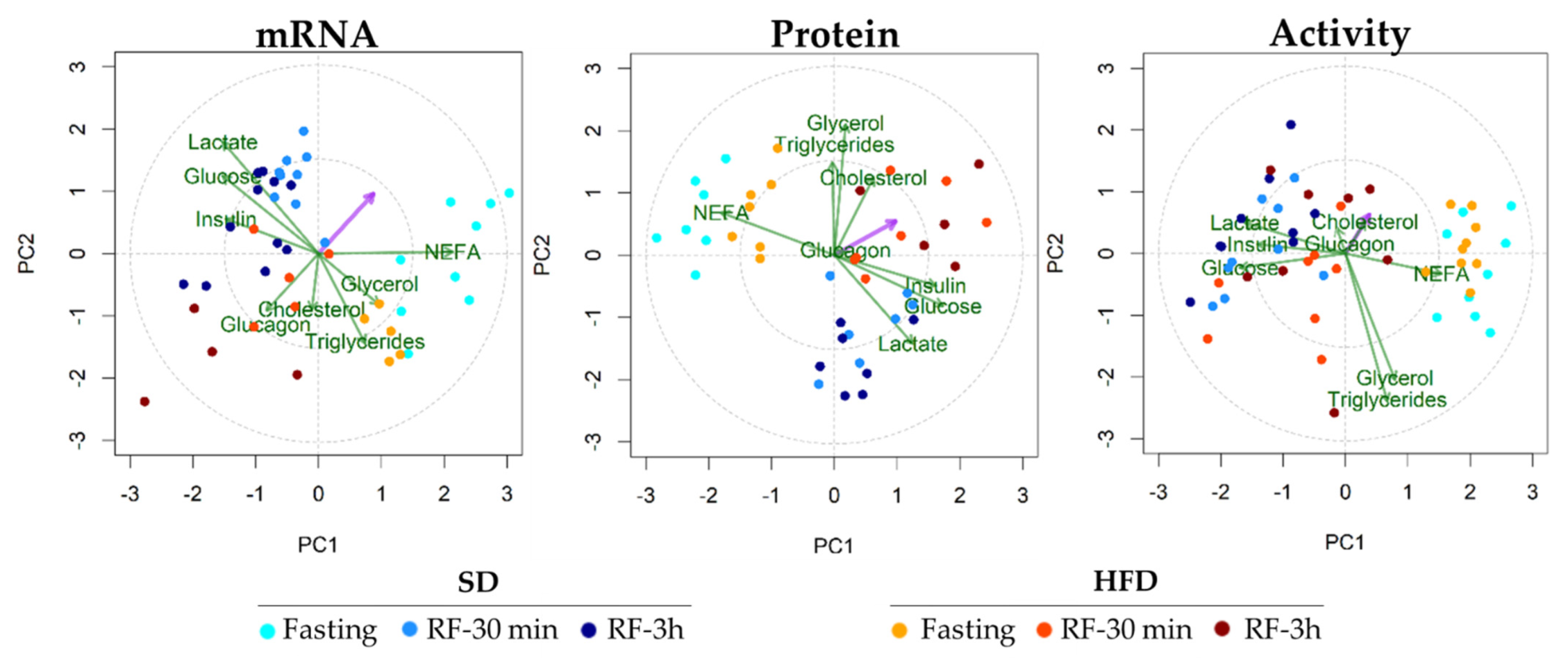
3.6. Multivariate Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Body Weight (g) | Glucose (mg/dL) | NEFA (mmol/L) | Cholesterol (mg/dL) | Triglycerides (mg/dL) | Glycerol (mg/L) | Lactate (mmol/L) | Insulin (ng/mL) | Glucagon (pmol/L) |
|---|---|---|---|---|---|---|---|---|---|
| Ide mRNA levels (HFD) | 0.0393 | 0.0685 | 0.1250 | 0.0662 | 0.0069 | 0.0010 | 0.1422 | 0.1600 | 0.1251 |
| p values | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 0.0399 | 0.031 | n.s. |
| Variables | Body Weight (g) | Glucose (mg/dL) | NEFA (mmol/L) | Cholesterol (mg/dL) | Triglycerides (mg/dL) | Glycerol (mg/L) | Lactate (mmol/L) | Insulin (ng/mL) | Glucagon (pmol/L) |
|---|---|---|---|---|---|---|---|---|---|
| IDE protein levels fasting (HFD) | 0.0382 | 0.0405 | 0.3840 | 0.0372 | 0.2374 | 0.0428 | 0.02958 | 0.1451 | 0.0737 |
| p values | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. |
| Variable | Body Weight (g) | Glucose (mg/dL) | NEFA (mmol/L) | Cholesterol (mg/dL) | Triglycerides (mg/dL) | Glycerol (mg/L) | Lactate (mmol/L) | Insulin (ng/mL) | Glucagon (pmol/L) |
|---|---|---|---|---|---|---|---|---|---|
| Ide mRNA levels (SD) | 0.009 | 0.003 | 0.1703 | 0.023 | 0.006 | 0.08 | 0.1496 | 0.069 | 0.1022 |
| Ide mRNA levels (HFD) | 0.177 | 0. 221 | 0.052 | 0.1068 | 0.017 | 0.033 | 0.26 | 0.044 | 0.044 |
| Ide mRNA levels (SD and HFD) | 0.268 | 0.049 | 0.242 | 0.115 | 0.021 | 0.085 | 0.020 | 0.187 | 0.266 |
| p values | n.s. | n.s. | 0.01 | n.s. | n.s. | n.s. | 0.03 | n.s. | n.s. |
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | |
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. |
References
- Brown, J.C.; Carson, T.L.; Thompson, H.J.; Agurs-Collins, T. The Triple Health Threat of Diabetes, Obesity, and Cancer—Epidemiology, Disparities, Mechanisms, and Interventions. Obesity 2021, 29, 954–959. [Google Scholar] [CrossRef]
- Kuo, W.L.; Montag, A.G.; Rosner, M.R. Insulin-degrading enzyme is differentially expressed and developmentally regulated in various rat tissues. Endocrinology 1993, 132, 604–611. [Google Scholar] [CrossRef]
- Mirsky, I.A.; Broh-Kahn, R.H. The inactivation of insulin by tissue extracts; the distribution and properties of insulin inactivating extracts. Arch. Biochem. 1949, 20, 1–9. [Google Scholar] [PubMed]
- González-Casimiro, C.M.; Merino, B.; Casanueva-Álvarez, E.; Postigo-Casado, T.; Cámara-Torres, P.; Fernández-Díaz, C.M.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Modulation of Insulin Sensitivity by Insulin-Degrading Enzyme. Biomedicines 2021, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Castell-Auví, A.; Cedó, L.; Pallarès, V.; Blay, M.; Ardévol, A.; Pinent, M. The effects of a cafeteria diet on insulin production and clearance in rats. Br. J. Nutr. 2012, 108, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandimarti, P.; Costa-Júnior, J.M.; Ferreira, S.M.; Protzek, A.O.; Santos, G.J.; Carneiro, E.M.; Boschero, A.C.; Rezende, L.F. Cafeteria diet inhibits insulin clearance by reduced insulin-degrading enzyme expression and mRNA splicing. J. Endocrinol. 2013, 219, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Ke, B.; Zhao, Z.; Ye, X.; Gao, Z.; Ye, J. Regulation of insulin degrading enzyme activity by obesity-associated factors and pioglitazone in liver of diet-induced obese mice. PLoS ONE 2014, 9, e95399. [Google Scholar] [CrossRef] [Green Version]
- Kurauti, M.A.; Costa-Júnior, J.M.; Ferreira, S.M.; Dos Santos, G.J.; Protzek, A.O.; Nardelli, T.R.; de Rezende, L.F.; Boschero, A.C. Acute exercise restores insulin clearance in diet-induced obese mice. J. Endocrinol. 2016, 229, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Elseweidy, M.M.; Amin, R.S.; Atteia, H.H.; Ali, M.A. Vitamin D3 intake as regulator of insulin degrading enzyme and insulin receptor phosphorylation in diabetic rats. Biomed. Pharmacother. 2017, 85, 155–159. [Google Scholar] [CrossRef]
- Vettorazzi, J.F.; Kurauti, M.A.; Soares, G.M.; Borck, P.C.; Ferreira, S.M.; Branco, R.C.S.; Michelone, L.S.L.; Boschero, A.C.; Junior, J.M.C.; Carneiro, E.M. Bile acid TUDCA improves insulin clearance by increasing the expression of insulin-degrading enzyme in the liver of obese mice. Sci. Rep. 2017, 7, 14876. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Rouse, M.; González-Mariscal, I.; Egan, J.M.; O’Connell, J.F. Dietary curcumin enhances insulin clearance in diet-induced obese mice via regulation of hepatic PI3K-AKT axis and IDE, and preservation of islet integrity. Nutr. Metab. 2019, 16, 48. [Google Scholar] [CrossRef]
- Borges, D.O.; Patarrão, R.S.; Ribeiro, R.T.; de Oliveira, R.M.; Duarte, N.; Belew, G.D.; Martins, M.; Andrade, R.; Costa, J.; Correia, I.; et al. Loss of postprandial insulin clearance control by Insulin-degrading enzyme drives dysmetabolism traits. Metab. Clin. Exp. 2021, 118, 154735. [Google Scholar] [CrossRef] [PubMed]
- Sofer, Y.; Nash, Y.; Osher, E.; Fursht, O.; Goldsmith, G.; Nahary, L.; Shaklai, S.; Tordjman, K.M.; Serebro, M.; Touati, E.B.; et al. Insulin-degrading enzyme higher in subjects with metabolic syndrome. Endocrine 2021, 71, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Fosam, A.; Sikder, S.; Abel, B.S.; Tella, S.H.; Walter, M.F.; Mari, A.; Muniyappa, R. Reduced Insulin Clearance and Insulin-Degrading Enzyme Activity Contribute to Hyperinsulinemia in African Americans. J. Clin. Endocrinol. Metab. 2020, 105, e1835–e1846. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, O.; von Loeffelholz, C.; Ilkavets, I.; Sticht, C.; Zhuk, S.; Murahovschi, V.; Lukowski, S.; Döcke, S.; Kriebel, J.; Gala, T.D.L.H.; et al. Modulation of insulin degrading enzyme activity and liver cell proliferation. Cell Cycle 2015, 14, 2293–2300. [Google Scholar] [CrossRef] [Green Version]
- Villa-Pérez, P.; Merino, B.; Fernández-Díaz, C.M.; Cidad, P.; Lobatón, C.D.; Moreno, A.; Muturi, H.T.; Ghadieh, H.E.; Najjar, S.M.; Leissring, M.A.; et al. Liver-specific ablation of insulin-degrading enzyme causes hepatic insulin resistance and glucose intolerance, without affecting insulin clearance in mice. Metab. Clin. Exp. 2018, 88, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Merino, B.; Fernández-Díaz, C.M.; Parrado-Fernández, C.; González-Casimiro, C.M.; Postigo-Casado, T.; Lobatón, C.D.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Hepatic insulin-degrading enzyme regulates glucose and insulin homeostasis in diet-induced obese mice. Metab. Clin. Exp. 2020, 113, 154352. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Fernández-Díaz, C.M.; Escobar-Curbelo, L.; López-Acosta, J.F.; Lobatón, C.D.; Moreno, A.; Sanz-Ortega, J.; Perdomo, G.; Cózar-Castellano, I. Insulin degrading enzyme is up-regulated in pancreatic β cells by insulin treatment. Histol. Histopathol. 2018, 33, 1167–1180. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2019. Available online: https://www.R-project.org/ (accessed on 13 June 2021).
- Mevik, B.-H.; Wehrens, R.; Liland, K.H.; Hiemstra, P. PLS: Partial Least Squares and Principal Component Regression. R Package Version 2.7-3. Available online: https://CRAN.R-project.org/package=pls (accessed on 13 June 2021).
- Martens, H.; Naes, T. Multivariate Calibration; Wiley: New York, NY, USA, 1989; Volume 1. [Google Scholar]
- Crispell, J. BasicPlotteR: A Collection of Functions to Help with Base R Plotting. R Package Version 0.0.0.9000. Available online: https://github.com/JosephCrispell/basicPlotteR (accessed on 13 June 2021).
- Lemon, J. Plotrix: A package in the red light district of R. R-News 2006, 6, 8–12. [Google Scholar]
- Farris, W.; Leissring, M.A.; Hemming, M.L.; Chang, A.Y.; Selkoe, D.J. Alternative splicing of human insulin-degrading enzyme yields a novel isoform with a decreased ability to degrade insulin and amyloid beta-protein. Biochemistry 2005, 44, 6513–6525. [Google Scholar] [CrossRef]
- Li, F.; Yang, J.; Jones, J.E.; Villar, V.A.; Yu, P.; Armando, I.; Felder, R.A.; Jose, P.A. Sorting nexin 5 and dopamine d1 receptor regulate the expression of the insulin receptor in human renal proximal tubule cells. Endocrinology 2015, 156, 2211–2221. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yang, J.; Villar, V.A.M.; Asico, L.D.; Ma, X.; Armando, I.; Sanada, H.; Yoneda, M.; Felder, R.A.; Jose, P.A.; et al. Loss of renal SNX5 results in impaired IDE activity and insulin resistance in mice. Diabetologia 2018, 61, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrén, B.; Simonsson, E.; Scheurink, A.J.; Mulder, H.; Myrsén, U.; Sundler, F. Dissociated insulinotropic sensitivity to glucose and carbachol in high-fat diet-induced insulin resistance in C57BL/6J mice. Metab. Clin. Exp. 1997, 46, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Conarello, S.L.; Jiang, G.; Mu, J.; Li, Z.; Woods, J.; Zycband, E.; Ronan, J.; Liu, F.; Roy, R.S.; Zhu, L.; et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 2007, 50, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Ellingsgaard, H.; Ehses, J.A.; Hammar, E.B.; Van Lommel, L.; Quintens, R.; Martens, G.; Kerr-Conte, J.; Pattou, F.; Berney, T.; Pipeleers, D.; et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc. Natl. Acad. Sci. USA 2008, 105, 13163–13168. [Google Scholar] [CrossRef] [Green Version]
- Merino, B.; Alonso-Magdalena, P.; Lluesma, M.; Ñeco, P.; Gonzalez, A.; Marroquí, L.; García-Arévalo, M.; Nadal, A.; Quesada, I. Pancreatic alpha-cells from female mice undergo morphofunctional changes during compensatory adaptations of the endocrine pancreas to diet-induced obesity. Sci. Rep. 2015, 5, 11622. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Jiang, G.; Brady, E.; Dallas-Yang, Q.; Liu, F.; Woods, J.; Zycband, E.; Wright, M.; Li, Z.; Lu, K.; et al. Chronic treatment with a glucagon receptor antagonist lowers glucose and moderately raises circulating glucagon and glucagon-like peptide 1 without severe alpha cell hypertrophy in diet-induced obese mice. Diabetologia 2011, 54, 2381–2391. [Google Scholar] [CrossRef]
- Winzell, M.S.; Brand, C.L.; Wierup, N.; Sidelmann, U.G.; Sundler, F.; Nishimura, E.; Ahrén, B. Glucagon receptor antagonism improves islet function in mice with insulin resistance induced by a high-fat diet. Diabetologia 2007, 50, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Item, F.; Konrad, D. Visceral fat and metabolic inflammation: The portal theory revisited. Obes. Rev. 2012, 13, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Hamel, F.G.; Upward, J.L.; Bennett, R.G. In vitro inhibition of insulin-degrading enzyme by long-chain fatty acids and their coenzyme A thioesters. Endocrinology 2003, 144, 2404–2408. [Google Scholar] [CrossRef] [Green Version]
- Svedberg, J.; Björntorp, P.; Smith, U.; Lönnroth, P. Free-fatty acid inhibition of insulin binding, degradation, and action in isolated rat hepatocytes. Diabetes 1990, 39, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Sonne, O. Increased inhibitory potency of free fatty acid-poor albumin on the released and activity of insulin-degrading enzymes from isolated rat adipocytes and hepatocytes. Anal. Biochem. 1985, 151, 109–117. [Google Scholar] [CrossRef]
- Pivovarova, O.; Gögebakan, O.; Pfeiffer, A.F.; Rudovich, N. Glucose inhibits the insulin-induced activation of the insulin-degrading enzyme in HepG2 cells. Diabetologia 2009, 52, 1656–1664. [Google Scholar] [CrossRef] [Green Version]
- Consoli, A.; Nurjahan, N.; Gerich, J.E.; Mandarino, L.J. Skeletal muscle is a major site of lactate uptake and release during hyperinsulinemia. Metab. Clin. Exp. 1992, 41, 176–179. [Google Scholar] [CrossRef]
- Consoli, A.; Nurjhan, N.; Reilly, J.J., Jr.; Bier, D.M.; Gerich, J.E. Contribution of liver and skeletal muscle to alanine and lactate metabolism in humans. Am. J. Physiol. 1990, 259, E677–E684. [Google Scholar] [CrossRef]
- Van Hall, G. Lactate kinetics in human tissues at rest and during exercise. Acta Physiol. 2010, 199, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, E.; Leech, T. Carbohydrate metabolism. In Functional Biochemistry in Health and Disease; Wiley-Blackwell: Oxford, UK, 2009; pp. 97–127. [Google Scholar]
- Kreisberg, R.A.; Pennington, L.F.; Boshell, B.R. Lactate turnover and gluconeogenesis in normal and obese humans: Effect of starvation. Diabetes 1970, 19, 53–63. [Google Scholar] [CrossRef]
- Andres, R.; Cader, G.; Zierler, K.L. The quantitatively minor role of carbohydrate in oxidative metabolism by skeletal muscle in intact man in the basal state; measurements of oxygen and glucose uptake and carbon dioxide and lactate production in the forearm. J. Clin. Investig. 1956, 35, 671–682. [Google Scholar] [CrossRef] [Green Version]
- Shulman, G.I.; Rossetti, L.; Rothman, D.L.; Blair, J.B.; Smith, D. Quantitative analysis of glycogen repletion by nuclear magnetic resonance spectroscopy in the conscious rat. J. Clin. Investig. 1987, 80, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; Mokan, M.; Simoneau, J.A.; Mandarino, L.J. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J. Clin. Investig. 1993, 92, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.E.; Mandarino, L.J. Fuel selection in human skeletal muscle in insulin resistance: A reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storlien, L.; Oakes, N.D.; Kelley, D.E. Metabolic flexibility. Proc. Nutr. Soc. 2004, 63, 363–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.E. Skeletal muscle fat oxidation: Timing and flexibility are everything. J. Clin. Investig. 2005, 115, 1699–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, D.; Mitrakou, A.; Marsh, H.; Schwenk, F.; Benn, J.; Sonnenberg, G.; Arcangeli, M.; Aoki, T.; Sorensen, J.; Berger, M. Skeletal muscle glycolysis, oxidation, and storage of an oral glucose load. J. Clin. Investig. 1988, 81, 1563–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidossis, L.S.; Wolfe, R.R. Glucose and insulin-induced inhibition of fatty acid oxidation: The glucose-fatty acid cycle reversed. Am. J. Physiol. 1996, 270, E733–E738. [Google Scholar] [CrossRef]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, Y.; Takeda, R.; Kano, Y.; Hoshino, D. Short-Term Calorie Restriction Maintains Plasma Insulin Concentrations along with a Reduction in Hepatic Insulin-Degrading Enzyme Levels in db/db Mice. Nutrients 2021, 13, 1190. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I.; Rothman, D.L.; Jue, T.; Stein, P.; DeFronzo, R.A.; Shulman, R.G. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N. Engl. J. Med. 1990, 322, 223–228. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| SD | HFD | |||||
|---|---|---|---|---|---|---|
| Fasting | Refeeding 30-min. | Refeeding 3-h | Fasting | Refeeding 30-min. | Refeeding 3-h | |
| Animals (n) | 9 | 10 | 10 | 10 | 11 | 11 |
| Animal Weight (g) | 19.94 ± 0.33 | 21.37 ± 0.43 | 21.41 ± 0.32 | 25.85 ± 0.34 $ | 25.73 ± 0.55 $ | 25.46 ± 0.37 $ |
| CI (kcal/kg) | ― | 96.42 ± 4.02 # | 258.25 ± 15.55 # | ― | 87.77 ± 10.20 # | 192.11 ± 16.70 # |
| CI rate (kcal/kg/min) | ― | 3.21 ± 0.13 # | 1.43 ± 0.09 # | ― | 2.93 ± 0.34 # | 1.07 ± 0.09 # |
| Glucose (mg/dL) | 96.33 ± 3.87 | 214.80 ± 8.17 *# | 173.40 ± 6.30 *# | 111.60 ± 5.26 | 218.82 ± 8.97 * | 174.55 ± 4.89 * |
| Insulin (ng/mL) | 0.09 ± 0.01 | 1.13 ± 0.12 * | 1.09 ± 0.17 * | 0.13 ± 0.00 $ | 0.92 ± 0.15 * | 1.55 ± 0.25 *$ |
| Glucagon (pmol/L) | 33.51 ± 8.75 | 12.38 ± 1.27 | 46.97 ± 19.37 | 8.02 ± 1.07 $ | 13.33 ± 1.94 | 30.67 ± 12.12 * |
| Cholesterol (mg/dL) | 61.00 ± 5.06 | 69.45 ± 3.76 | 59.91 ± 5.38 | 93.77 ± 3.50 | 91.79 ± 6.52 | 106.18 ±5.17 $ |
| Triglycerides (mg/dL) | 88.12 ± 7.40 | 71.63 ± 5.40 | 69.40 ± 5.50 | 94.59 ± 3.41 | 89.31 ± 5.33 | 91.74 ± 5.63 |
| NEFA (mmol/L) | 1.75 ± 0.12 | 0.65 ± 0.03 | 0.41 ± 0.05 * | 1.00 ± 0.03 | 0.37 ± 0.02 * | 0.30 ± 0.02 |
| Glycerol (mg/L) | 35.57 ± 1.43 | 26.06 ± 0.88 | 17.72 ± 1.26 * | 37.62 ± 2.36 | 38.96 ± 2.10 $ | 35.14 ± 3.54 $ |
| Lactate (mmol/L) | 2.62 ± 0.33 | 6.23 ± 0.28 * | 6.52 ± 0.24 * | 1.98 ± 0.08 | 4.15 ± 0.33 $ | 4.12 ± 0.19 $ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Casimiro, C.M.; Cámara-Torres, P.; Merino, B.; Diez-Hermano, S.; Postigo-Casado, T.; Leissring, M.A.; Cózar-Castellano, I.; Perdomo, G. Effects of Fasting and Feeding on Transcriptional and Posttranscriptional Regulation of Insulin-Degrading Enzyme in Mice. Cells 2021, 10, 2446. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092446
González-Casimiro CM, Cámara-Torres P, Merino B, Diez-Hermano S, Postigo-Casado T, Leissring MA, Cózar-Castellano I, Perdomo G. Effects of Fasting and Feeding on Transcriptional and Posttranscriptional Regulation of Insulin-Degrading Enzyme in Mice. Cells. 2021; 10(9):2446. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092446
Chicago/Turabian StyleGonzález-Casimiro, Carlos M., Patricia Cámara-Torres, Beatriz Merino, Sergio Diez-Hermano, Tamara Postigo-Casado, Malcolm A. Leissring, Irene Cózar-Castellano, and Germán Perdomo. 2021. "Effects of Fasting and Feeding on Transcriptional and Posttranscriptional Regulation of Insulin-Degrading Enzyme in Mice" Cells 10, no. 9: 2446. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092446