
Generation of HepG2 Cells with High Expression of Multiple Drug-Metabolizing Enzymes for Drug Discovery Research Using a PITCh System

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. HepG2 Cells
2.3. Primary Human Hepatocytes
2.4. Vector Construction
2.5. Generation of CYPs–UGT1A1 KI-HepG2 Cells
2.6. Real-Time RT-PCR
2.7. HPLC
2.8. Cell Viability Tests
2.9. Statistical Analysis
3. Results
3.1. Generation of CYPs–UGT1A1 KI-HepG2 Cells
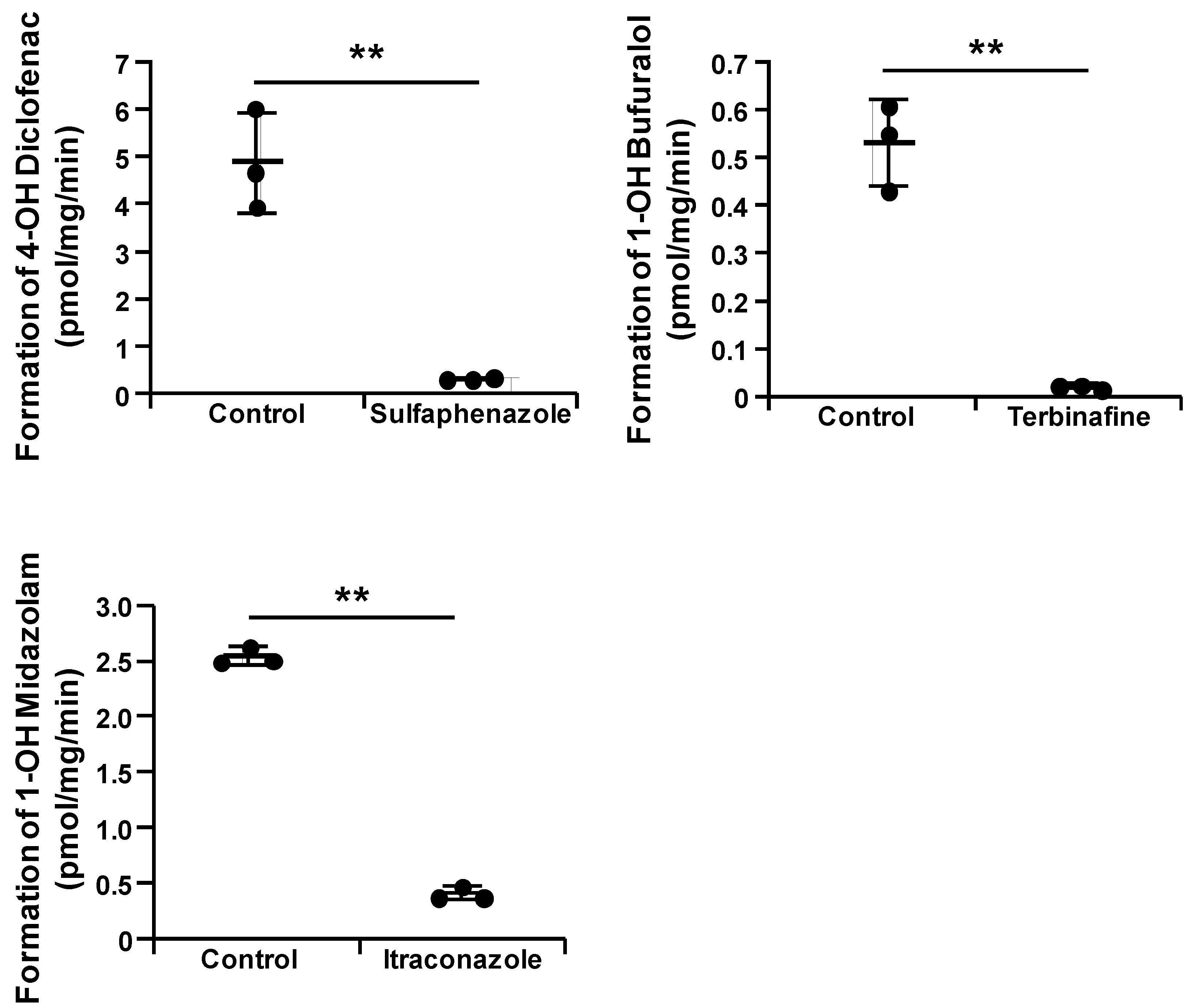
3.2. Drug-Metabolizing Activity Evaluation of the CYPs–UGT1A1 KI-HepG2 Cells
3.3. Prediction of Drug-Induced Liver Injury Using CYPs–UGT1A1 KI-HepG2 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, S.; Feng, S.; Wu, Z.; He, T.; Ma, C.; Peng, Z.; Tian, E.; Pan, G. Functional Proliferating Human Hepatocytes: In Vitro Hepatocyte Model for Drug Metabolism, Excretion, and Toxicity. Drug Metab. Dispos. 2021, 49, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; Björnsson, E.S. Drug-Induced Liver Injury—Types and Phenotypes. N. Engl. J. Med. 2019, 381, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Lu, A.Y. Inhibition and induction of cytochrome P450 and the clinical implications. Clin. Pharmacokinet. 1998, 35, 361–390. [Google Scholar] [CrossRef]
- Agrawal, V.; Choi, J.H.; Giacomini, K.M.; Miller, W.L. Substrate-specific modulation of CYP3A4 activity by genetic variants of cytochrome P450 oxidoreductase. Pharmacogenet. Genom. 2010, 20, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Weaver, R.J.; Blomme, E.A.; Chadwick, A.E.; Copple, I.M.; Gerets, H.H.J.; Goldring, C.E.; Guillouzo, A.; Hewitt, P.G.; Ingelman-Sundberg, M.; Jensen, K.G.; et al. Managing the challenge of drug-induced liver injury: A roadmap for the development and deployment of preclinical predictive models. Nat. Rev. Drug Discov. 2020, 19, 131–148. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Sasaki, Y.; Terasaki, N.; Kawataki, T.; Takekawa, K.; Iwase, Y.; Shimizu, T.; Sanoh, S.; Ohta, S. Comparison of Drug Metabolism and Its Related Hepatotoxic Effects in HepaRG, Cryopreserved Human Hepatocytes, and HepG2 Cell Cultures. Biol. Pharm. Bull. 2018, 41, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Brandon, E.F.A.; Raap, C.D.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H.M. An update on in vitro test methods in human hepatic drug biotransformation research: Pros and cons. Toxicol. Appl. Pharmacol. 2003, 189, 233–246. [Google Scholar] [CrossRef]
- Schulz, C.; Kammerer, S.; Küpper, J.-H. NADPH-cytochrome P450 reductase expression and enzymatic activity in primary-like human hepatocytes and HepG2 cells for in vitro biotransformation studies. Clin. Hemorheol. Microcirc. 2019, 73, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.C.; Logan, C.Y.; Fish, M.; Anbarchian, T.; Aguisanda, F.; Álvarez-Varela, A.; Wu, P.; Jin, Y.; Zhu, J.; Li, B.; et al. Inflammatory Cytokine TNFα Promotes the Long-Term Expansion of Primary Hepatocytes in 3D Culture. Cell 2018, 175, 1607–1619.e15. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, D.; Artegiani, B.; Hu, H.; Chuva de Sousa Lopes, S.; Clevers, H. Establishment of human fetal hepatocyte organoids and CRISPR-Cas9-based gene knockin and knockout in organoid cultures from human liver. Nat. Protoc. 2021, 16, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Westerink, W.M.A.; Schoonen, W.G.E.J. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 2007, 21, 1581–1591. [Google Scholar] [CrossRef]
- Westerink, W.M.A.; Schoonen, W.G.E.J. Phase II enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 2007, 21, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, Q.; Li, X.; Li, D.; Mei, N.; Ning, B.; Puig, M.; Ren, Z.; Tolleson, W.H.; Guo, L. Characterization of cytochrome P450s (CYP)-overexpressing HepG2 cells for assessing drug and chemical-induced liver toxicity. J. Environ. Sci. Health Part C Toxicol. Carcinog. 2021, 39, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Mizuguchi, H. Generation of human pluripotent stem cell-derived hepatocyte-like cells for drug toxicity screening. Drug Metab. Pharmacokinet. 2017, 32, 12–20. [Google Scholar] [CrossRef]
- Takayama, K.; Morisaki, Y.; Kuno, S.; Nagamoto, Y.; Harada, K.; Furukawa, N.; Ohtaka, M.; Nishimura, K.; Imagawa, K.; Sakurai, F.; et al. Prediction of interindividual differences in hepatic functions and drug sensitivity by using human iPS-derived hepatocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 16772–16777. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, S.; Yamashita, T.; Igai, K.; Harada, K.; Toba, Y.; Hirata, K.; Takayama, K.; Mizuguchi, H. Modeling of Hepatic Drug Metabolism and Responses in CYP2C19 Poor Metabolizer Using Genetically Manipulated Human iPS cells. Drug Metab. Dispos. 2019, 47, 632–638. [Google Scholar] [CrossRef]
- Baxter, M.; Withey, S.; Harrison, S.; Segeritz, C.-P.; Zhang, F.; Atkinson-Dell, R.; Rowe, C.; Gerrard, D.T.; Sison-Young, R.; Jenkins, R.; et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015, 62, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, N.A.; Meille, C.; Fowler, S.; Klammers, F.; Ekiciler, A.; Molitor, B.; Simon, S.; Walter, I.; McGinnis, C.; Walther, J.; et al. Metabolic Profiling of Human Long-Term Liver Models and Hepatic Clearance Predictions from In Vitro Data Using Nonlinear Mixed-Effects Modeling. AAPS J. 2017, 19, 534–550. [Google Scholar] [CrossRef] [PubMed]
- Nghiem-Rao, T.H.; Pfeifer, C.; Asuncion, M.; Nord, J.; Schill, D.; Pulakanti, K.; Patel, S.B.; Cirillo, L.A.; Rao, S. Human induced pluripotent stem cell derived hepatocytes provide insights on parenteral nutrition associated cholestasis in the immature liver. Sci. Rep. 2021, 11, 12386. [Google Scholar] [CrossRef] [PubMed]
- Negoro, R.; Yamada, N.; Watanabe, K.; Kono, Y.; Fujita, T. Generation of Caco-2 cells stably expressing CYP3A4·POR·UGT1A1 and CYP3A4·POR·UGT1A1*6 using a PITCh system. Arch. Toxicol. 2022, 96, 499–510. [Google Scholar] [CrossRef]
- Deguchi, S.; Tsuda, M.; Kosugi, K.; Sakamoto, A.; Mimura, N.; Negoro, R.; Sano, E.; Nobe, T.; Maeda, K.; Kusuhara, H.; et al. Usability of Polydimethylsiloxane-Based Microfluidic Devices in Pharmaceutical Research Using Human Hepatocytes. ACS Biomater. Sci. Eng. 2021, 7, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Sano, E.; Deguchi, S.; Matsuoka, N.; Tsuda, M.; Wang, M.; Kosugi, K.; Mori, C.; Yagi, K.; Wada, A.; Yamasaki, S.; et al. Generation of Tetrafluoroethylene-Propylene Elastomer-Based Microfluidic Devices for Drug Toxicity and Metabolism Studies. ACS Omega 2021, 6, 24859–24865. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.I.T.; Yamamoto, T. MMEJ-Assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [Google Scholar] [CrossRef]
- Shirasaka, Y.; Chang, S.-Y.; Grubb, M.F.; Peng, C.-C.; Thummel, K.E.; Isoherranen, N.; Rodrigues, A.D. Effect of CYP3A5 expression on the inhibition of CYP3A-catalyzed drug metabolism: Impact on modeling CYP3A-mediated drug-drug interactions. Drug Metab. Dispos. 2013, 41, 1566–1574. [Google Scholar] [CrossRef] [Green Version]
- McGinnity, D.F.; Tucker, J.; Trigg, S.; Riley, R.J. Prediction of CYP2C9-mediated drug-drug interactions: A comparison using data from recombinant enzymes and human hepatocytes. Drug Metab. Dispos. 2005, 33, 1700–1707. [Google Scholar] [CrossRef] [Green Version]
- Bell, L.; Bickford, S.; Nguyen, P.H.; Wang, J.; He, T.; Zhang, B.; Friche, Y.; Zimmerlin, A.; Urban, L.; Bojanic, D. Evaluation of fluorescence- and mass spectrometry-based CYP inhibition assays for use in drug discovery. J. Biomol. Screen. 2008, 13, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosomi, H.; Fukami, T.; Iwamura, A.; Nakajima, M.; Yokoi, T. Development of a highly sensitive cytotoxicity assay system for CYP3A4-mediated metabolic activation. Drug Metab. Dispos. 2011, 39, 1388–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrecht, S.; Pfeifer, N.; Herzog, N.; Katzenberger, N.; Schulz, C.; Kammerer, S.; Küpper, J.-H. HepG2-1A2 C2 and C7: Lentivirus vector-mediated stable and functional overexpression of cytochrome P450 1A2 in human hepatoblastoma cells. Toxicol. Lett. 2020, 319, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, S.; Ikemoto, K.; Takahashi, J.; Miki, H.; Namba, M.; Asahi, S. Establishment of the transformants expressing human cytochrome P450 subtypes in HepG2, and their applications on drug metabolism and toxicology. Toxicol. Vitr. 2001, 15, 245–256. [Google Scholar] [CrossRef]
- Xuan, J.; Chen, S.; Ning, B.; Tolleson, W.H.; Guo, L. Development of HepG2-derived cells expressing cytochrome P450s for assessing metabolism-associated drug-induced liver toxicity. Chem. Biol. Interact. 2016, 255, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Satoh, D.; Iwado, S.; Abe, S.; Kazuki, K.; Wakuri, S.; Oshimura, M.; Kazuki, Y. Establishment of a novel hepatocyte model that expresses four cytochrome P450 genes stably via mammalian-derived artificial chromosome for pharmacokinetics and toxicity studies. PLoS ONE 2017, 12, e0187072. [Google Scholar] [CrossRef] [Green Version]
- Otey, C.R.; Bandara, G.; Lalonde, J.; Takahashi, K.; Arnold, F.H. Preparation of human metabolites of propranolol using laboratory-evolved bacterial cytochromes P450. Biotechnol. Bioeng. 2006, 93, 494–499. [Google Scholar] [CrossRef]
- Walle, T.; Walle, U.K.; Olanoff, L.S. Quantitative account of propranolol metabolism in urine of normal man. Drug Metab. Dispos. 1985, 13, 204–209. [Google Scholar]
- Yoshimoto, K.; Echizen, H.; Chiba, K.; Tani, M.; Ishizaki, T. Identification of human CYP isoforms involved in the metabolism of propranolol enantiomers--N-desisopropylation is mediated mainly by CYP1A2. Br. J. Clin. Pharmacol. 1995, 39, 421–431. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Radominska-Pandya, A.; Tephly, T.R. Studies on the substrate specificity of human intestinal UDP-lucuronosyltransferases 1A8 and 1A10. Drug Metab. Dispos. 1999, 27, 1165–1170. [Google Scholar]
- Juvonen, R.O.; Heikkinen, A.T.; Kärkkäinen, O.; Jehangir, R.; Huuskonen, J.; Troberg, J.; Raunio, H.; Pentikäinen, O.T.; Finel, M. In vitro glucuronidation of 7-hydroxycoumarin derivatives in intestine and liver microsomes of Beagle dogs. Eur. J. Pharm. Sci. 2020, 141, 105118. [Google Scholar] [CrossRef] [PubMed]
- Juvonen, R.O.; Rauhamäki, S.; Kortet, S.; Niinivehmas, S.; Troberg, J.; Petsalo, A.; Huuskonen, J.; Raunio, H.; Finel, M.; Pentikäinen, O.T. Molecular Docking-Based Design and Development of a Highly Selective Probe Substrate for UDP-glucuronosyltransferase 1A10. Mol. Pharm. 2018, 15, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Genetic susceptibility to adverse effects of drugs and environmental toxicants: The role of the CYP family of enzymes. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2001, 482, 11–19. [Google Scholar] [CrossRef]
- Sai, K.; Saeki, M.; Saito, Y.; Ozawa, S.; Katori, N.; Jinno, H.; Hasegawa, R.; Kaniwa, N.; Sawada, J.; Komamura, K.; et al. UGT1A1 haplotypes associated with reduced glucuronidation and increased serum bilirubin in irinotecan-administered Japanese patients with cancer. Clin. Pharmacol. Ther. 2004, 75, 501–515. [Google Scholar] [CrossRef]
- Zahno, A.; Brecht, K.; Morand, R.; Maseneni, S.; Török, M.; Lindinger, P.W.; Krähenbühl, S. The role of CYP3A4 in amiodarone-associated toxicity on HepG2 cells. Biochem. Pharmacol. 2011, 81, 432–441. [Google Scholar] [CrossRef]
- Shayeganpour, A.; El-Kadi, A.O.S.; Brocks, D.R. Determination of the enzyme(s) involved in the metabolism of amiodarone in liver and intestine of rat: The contribution of cytochrome P450 3A isoforms. Drug Metab. Dispos. 2006, 34, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Kitagawara, Y.; Ohe, T.; Tachibana, K.; Takahashi, K.; Nakamura, S.; Mashino, T. Novel Bioactivation Pathway of Benzbromarone Mediated by Cytochrome P450. Drug Metab. Dispos. 2015, 43, 1303–1306. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Kajiwara, E.; Ishikawa, M.; Mimura, H.; Oka, H.; Ejiri, Y.; Hosoda, M.; Chiba, K. Cytotoxic effects of benzbromarone and its 1′-hydroxy metabolite in human hepatocarcinoma FLC4 cells cultured on micro-space cell culture plates. Drug Metab. Pharmacokinet. 2013, 28, 265–268. [Google Scholar] [CrossRef]
- James, L.P.; Mayeux, P.R.; Hinson, J.A. Acetaminophen-induced hepatotoxicity. Drug Metab. Dispos. 2003, 31, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. A history of the roles of cytochrome P450 enzymes in the toxicity of drugs. Toxicol. Res. 2021, 37, 1–23. [Google Scholar] [CrossRef]
- Deguchi, S.; Shintani, T.; Harada, K.; Okamoto, T.; Takemura, A.; Hirata, K.; Ito, K.; Takayama, K.; Mizuguchi, H. In Vitro Model for a Drug Assessment of Cytochrome P450 Family 3 Subfamily A Member 4 Substrates Using Human Induced Pluripotent Stem Cells and Genome Editing Technology. Hepatol. Commun. 2021, 5, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.; Hukkanen, J.; Raunio, H. Inhibition and induction of human cytochrome P450 enzymes: Current status. Arch. Toxicol. 2008, 82, 667–715. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negoro, R.; Tasaka, M.; Deguchi, S.; Takayama, K.; Fujita, T. Generation of HepG2 Cells with High Expression of Multiple Drug-Metabolizing Enzymes for Drug Discovery Research Using a PITCh System. Cells 2022, 11, 1677. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101677
Negoro R, Tasaka M, Deguchi S, Takayama K, Fujita T. Generation of HepG2 Cells with High Expression of Multiple Drug-Metabolizing Enzymes for Drug Discovery Research Using a PITCh System. Cells. 2022; 11(10):1677. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101677
Chicago/Turabian StyleNegoro, Ryosuke, Mitsuki Tasaka, Sayaka Deguchi, Kazuo Takayama, and Takuya Fujita. 2022. "Generation of HepG2 Cells with High Expression of Multiple Drug-Metabolizing Enzymes for Drug Discovery Research Using a PITCh System" Cells 11, no. 10: 1677. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101677