
Across Dimensions: Developing 2D and 3D Human iPSC-Based Models of Fragile X Syndrome


1
Neuroscience Graduate Program, Emory University, Atlanta, GA 30322, USA
2
MD/PhD Program, Emory University School of Medicine, Atlanta, GA 30322, USA
3
Genetics and Molecular Biology Graduate Program, Emory University, Atlanta, GA 30322, USA
4
Department of Psychiatry and Behavioral Sciences, Emory University School of Medicine, Atlanta, GA 30322, USA
5
Department of Cell Biology, Emory University School of Medicine, Atlanta, GA 30322, USA
6
Department of Human Genetics, Emory University School of Medicine, Atlanta, GA 30322, USA
*
Authors to whom correspondence should be addressed.
Cells 2022, 11(11), 1725; https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111725
Submission received: 10 March 2022
/
Revised: 11 May 2022
/
Accepted: 13 May 2022
/
Published: 24 May 2022
(This article belongs to the Special Issue Fragile X Syndrome: Molecular Mechanisms, Cellular and Animal Models, and Targeted Therapeutics)
Abstract
:Fragile X syndrome (FXS) is the most common inherited cause of intellectual disability and autism spectrum disorder. FXS is caused by a cytosine-guanine-guanine (CGG) trinucleotide repeat expansion in the untranslated region of the FMR1 gene leading to the functional loss of the gene’s protein product FMRP. Various animal models of FXS have provided substantial knowledge about the disorder. However, critical limitations exist in replicating the pathophysiological mechanisms. Human induced pluripotent stem cells (hiPSCs) provide a unique means of studying the features and processes of both normal and abnormal human neurodevelopment in large sample quantities in a controlled setting. Human iPSC-based models of FXS have offered a better understanding of FXS pathophysiology specific to humans. This review summarizes studies that have used hiPSC-based two-dimensional cellular models of FXS to reproduce the pathology, examine altered gene expression and translation, determine the functions and targets of FMRP, characterize the neurodevelopmental phenotypes and electrophysiological features, and, finally, to reactivate FMR1. We also provide an overview of the most recent studies using three-dimensional human brain organoids of FXS and end with a discussion of current limitations and future directions for FXS research using hiPSCs.
1. Introduction
1.1. Fragile X Syndrome
Fragile X syndrome (FXS) is a neurodevelopmental disorder characterized by developmental delay, intellectual disability, autism, attention-deficit/hyperactivity disorder, anxiety, and seizures [1,2,3]. FXS is the most common inherited cause of intellectual disability and the leading monogenic cause of autism spectrum disorder [1,3,4,5]. The study of FXS has provided, and will continue to provide, crucial insight to understanding the pathophysiology of the disorders and symptoms it encompasses. FXS is caused by an expansion of cytosine-guanine-guanine (CGG) trinucleotide repeats in the 5′-untranslated region (UTR) of the fragile X messenger ribonucleoprotein 1 (FMR1) gene on the X chromosome. When an individual has a full mutation with greater than 200 CGG repeats, the repeats themselves, as well as the promoter region of the FMR1 gene, are hypermethylated [6,7,8,9]. Due to abnormal methylation and histone modifications [10], FMR1 is silenced, leading to subsequent reduction or absence of its protein product fragile X messenger ribonucleoprotein (FMRP) [2,3,9,10,11,12]. FMRP is an RNA-binding protein that can modulate the translation of its mRNA targets involved in diverse essential cellular functions, including neurogenesis, axon and dendrite formation, synaptic plasticity, cytoskeletal organization, histone modification, and RNA transport [12,13,14,15]. The loss of FMRP has many consequences, with the clinical manifestation being FXS. Individuals with repeat numbers between 55 and 200 are referred to as premutation carriers and may develop other FMR1-associated disorders, such as fragile-X-associated tremor ataxia syndrome (FXTAS) for males and some females and fragile-X-associated primary ovarian insufficiency (FXPOI) for females [1,3,16,17,18].
Thirty years have passed since the discovery of the genetic and molecular mechanism of FXS [7,8,19], which has paved the way for understanding disorders similarly caused by nucleotide repeat expansions in other genes, including Huntington’s disease, Friedreich’s ataxia, myotonic dystrophy, spinocerebellar ataxias, and familial adult myoclonic epilepsies [20]. Active research on FXS has led to substantial progress in understanding the pathogenesis of the disorder. Nonetheless, there is not yet a gold standard treatment available for FXS. Various promising therapeutic targets for FXS have been discovered and tested in animal models of FXS, but most clinical trials have been unsuccessful [21,22,23,24]. Although animal models of FXS [25,26], including Drosophila, mouse, rat, and zebrafish, are invaluable in understanding the functions of FMRP, there are limitations because the pathogenic mechanism of FXS in humans, in which trinucleotide repeat expansions lead to gene silencing, does not apply to other species [6,12,27,28,29]. Knock-in mouse models with expanded CGG repeats do not reproduce the pathophysiological processes of FXS in humans [30,31]. Furthermore, recent studies have discovered that FMRP targets are species-specific: FMRP in humans may bind different RNAs than does FMRP in animal models of FXS [22,24]. Different neural cell types and brain organoids derived from human induced pluripotent stem cells (iPSCs) provide a unique means of studying the normal and pathological processes of neurodevelopment seen in FXS. In addition, using iPSC-derived cellular models of FXS enables the manipulation of various genes or proteins that are implicated to be altered in FXS. Examining the effects of these modulations provides a deeper insight into the pathogenesis, as well as highlighting promising therapeutic targets for FXS.
1.2. Pluripotent Stem Cells (PSCs) and Translational Applications
The advent of stem cell technology has opened a new avenue for studying human development and diseases in vitro. Unlike human postmortem tissues that are limited in quantity and represent only the end-stage status of a disease or human primary cells that can only be maintained for a limited time in vitro, human pluripotent stem cells (hPSCs), with the ability to self-renew indefinitely and the potential to differentiate into almost every cell type in the body, provide a renewable and inexhaustible resource for studying human development and diseases, as well as for testing therapeutic compounds and other treatment strategies.
There are two types of human pluripotent stem cells: human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs). hESCs are obtained primarily from the inner cell mass of supernumerary blastocysts created by in vitro fertilization (IVF), a method of assisted reproduction [32]. These pluripotent stem cells can be differentiated into various cell types of interest and used to investigate numerous processes of development and disease mechanisms. Many studies have used hESC-derived neural cells as a model for studying fragile X syndrome [28,29,33,34,35,36,37,38]. For example, Telias et al. (2013) investigated three male hESC lines (FXS-hESCs) that were obtained from spare IVF-derived embryos diagnosed with fragile X syndrome via preimplantation genetic diagnosis (PGD) [34]. They found that FMR1 was expressed in undifferentiated FXS-hESCs and was progressively inactivated through differentiation into neurons, consistent with the process that occurs in human FXS fetuses. Moreover, they observed aberrant expression patterns of key neural genes and delayed development of neural rosettes in FXS lines during early stages of differentiation, as well a significant shift towards the glial lineage during final stages of differentiation, leading to poor neuronal maturation and high gliogenic development in FXS lines [34].
Although such FXS-hESCs are advantageous in terms of recapitulating the in vivo process of FMR1 inactivation during differentiation, they are difficult to acquire given the limited resource of IVF-derived pre-diagnosed embryos. Therefore, an alternative approach has been taken utilizing gene editing techniques to create knockout (KO) of the FMR1 gene in healthy control hESC lines to model FXS in vitro. As an example, Li et al. (2020) made use of clustered regularly interspaced short palindromic repeats (CRISPR) technique to generate FMR1-KO hPSC lines (two FMR1-KO hESC lines and one FMR1-KO hiPSC line) for studying FMRP function [23]. Nevertheless, the use of hESCs raises ethical and legal issues and, therefore, is under strict regulations in many countries [39].
Human iPSCs, the other type of human pluripotent stem cells, have also been widely used for disease modeling since its first establishment in 2007 [40]. hiPSCs are derived from human somatic cells via reprograming with four transcription factors (Oct3/4, Sox2, Klf4, and c-Myc, also called Yamanaka factors) and are, therefore, more accessible than hESCs [40,41]. Since they carry the exact same genetic information as the donor, patient-derived hiPSCs would naturally contain the pathogenetic mutations and are, therefore, extremely helpful when differentiating into cell types of interest as models for studying disease mechanisms. Once a patient iPSC line(s) is established, it is also possible to perform large-scale high-throughput drug screening to identify effective drug treatments for the disease of interest. Furthermore, patient-specific iPSCs have promoted the development of regenerative medicine and cell therapies in a way that makes genetically corrected autologous cell transplantation possible and eliminates the possibilities of immune rejection from allogeneic cell transplantation [42,43,44]. Indeed, numerous studies have taken advantage of such disease models for their investigations.
Here in this review, we focus on compiling and summarizing research findings on hiPSC-based FXS models (Table 1; studies listed by section in the order of discussion). We also discuss the future directions of hiPSC-based studies on FXS.
2. Two-Dimensional (2D) Human iPSC-Based Cellular Models of FXS
Two-dimensional cellular models have several advantages, including the ability to culture large quantities of a relatively homogenous population of cells and maintain those cells at a particular stage of development. Using 2D cell models of disease, cell-type-specific phenotypes can be easily examined. Moreover, cells at certain developmental stages can be monitored and followed through stages of maturation and differentiation in a phase-locked fashion [45]. This section provides a review of the studies that have utilized two-dimensional iPSC-based cellular models of FXS and their findings.
2.1. Generation of Human iPSC-Derived Cellular Models of FXS
Early studies that generated iPSCs derived from primary cells of patients with FXS (hereafter referred to as FXS-iPSCs) found that FMR1 was inactive in iPSCs and its expression was not reactivated by reprograming the original somatic cells into iPSCs [46,47], nor by differentiation into neurons [47] (Figure 1A). FMR1 was confirmed to remain silent in iPSCs derived from dermal fibroblasts from a pediatric FXS patient, those from an adult patient of FXS, as well as lung fibroblasts from a fetus diagnosed with FXS [46]. The FMR1 locus was methylated in FXS-iPSCs and contained histone modifications associated with a heterochromatin state of epigenetic markers, including increased H3K9 methylation and reduced H3 acetylation and H3K4 methylation [46]. The absence of FMRP expression further verified the silencing of FMR1 in FXS-iPSCs [46,47,48].
While most molecular mechanisms of FXS are maintained in iPSC-based models of the disorder, a few studies have reported discrepancies in primary cells and iPSCs with fragile X full mutations. One study, which examined the level of 5-hydroxymethylcytosine (5hmC), an intermediate epigenetic marker for DNA demethylation, at the FMR1 promoter region, reported differential findings in primary neurons of FXS patients and iPSCs derived from FXS patients [49]. 5hmC was found to be enriched at the FMR1 promoter in primary neurons isolated from postmortem brain tissues of FXS patients compared to controls but not in FXS-iPSCs, FXS-iPSC-derived neurons, or non-neuronal cell lines collected from FXS patients [49].
Another study suggested that the reprograming process could introduce instability of the expanded CGG repeats in that the iPSCs generated from fibroblasts of FXS patients had variable ranges of CGG repeat lengths, although all above the normal repeat size, that could differ from those of the original fibroblast population [47]. Furthermore, FMR1 was found to be methylated in subsets of iPSCs generated from fibroblasts of individuals with an unmethylated full mutation (UFM), supporting that the reprograming process can direct silencing of FMR1 with a full mutation [50,51]. The iPSC lines from UFM individuals were found to have a higher CGG repeat size threshold for FMR1 silencing—greater than 400—compared to the threshold of 200 repeats seen in FXS patients [51]. These findings emphasize the importance of determining CGG repeat lengths and FMR1 silencing status in any FXS-iPSC cell line.
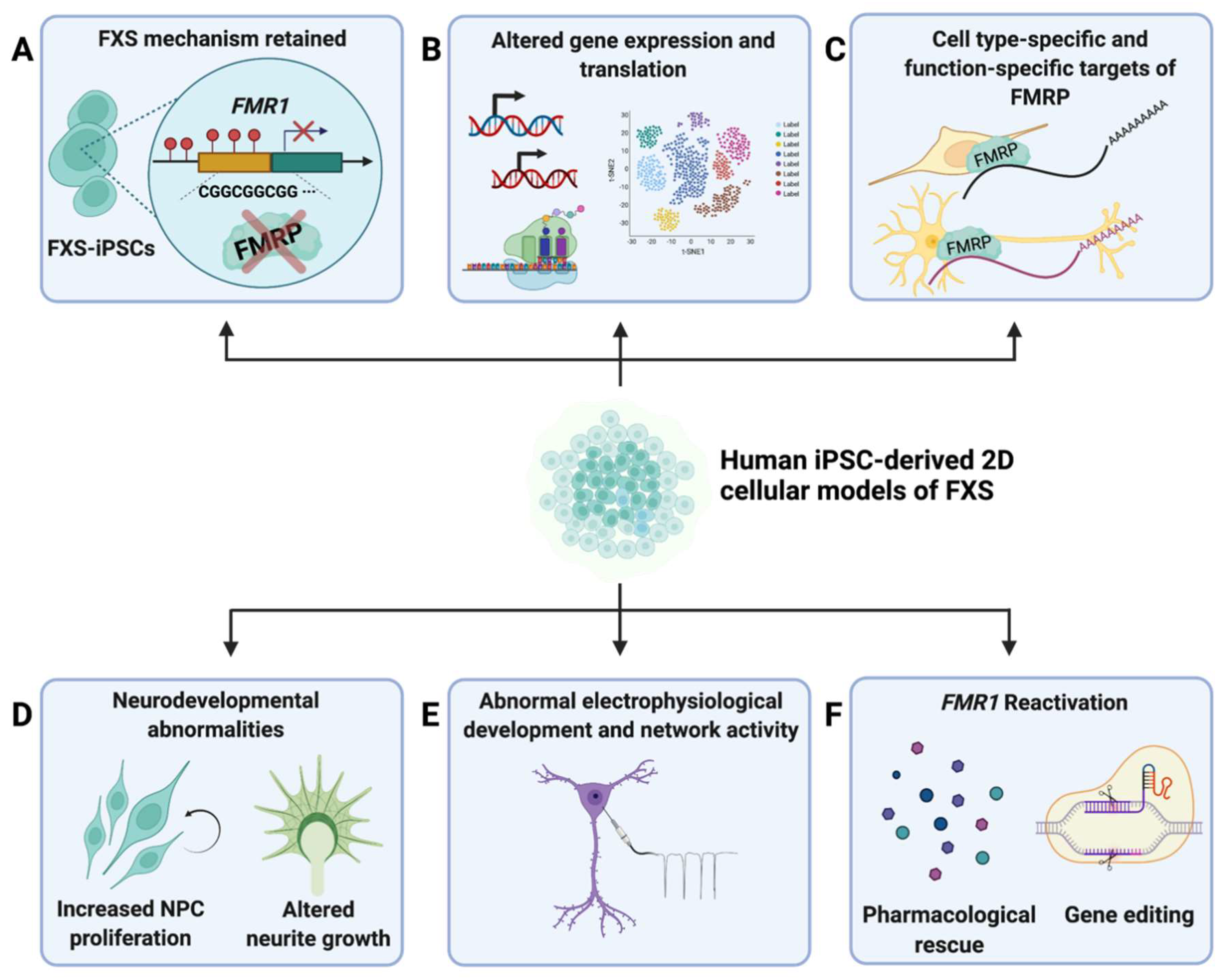
Figure 1.
Summary of major findings from 2D FXS−iPSC−derived models. (A) Studies that have utilized 2D hiPSC-derived models of FXS have found that the fundamental molecular mechanisms of FXS pathology is retained in iPSCs derived from primary cells of FXS patients (FXS-iPSCs) [46,47]. (B) FXS-iPSCs and derived neural cells showed altered gene expression and translation compared to controls [21,52,53,54,55,56,57]. (C) Studies of targets and functions of FMRP showed that there are cell-type-specific [23] and function-specific [58] targets of FMRP [23,58]. (D) Neurodevelopmental abnormalities, including increased NPC proliferation [21,23,53] and altered neurite growth [47,48,53,55,59], were characterized in iPSC-derived models of FXS. (E) FXS-iPSC-derived neural cells exhibited electrophysiological abnormalities [24,59,60,61,62,63]. (F) Pharmacological rescue [64,65,66,67,68,69] and gene editing [70,71,72,73] are two major methods of FMR1 reactivation that have been explored.
Figure 1.
Summary of major findings from 2D FXS−iPSC−derived models. (A) Studies that have utilized 2D hiPSC-derived models of FXS have found that the fundamental molecular mechanisms of FXS pathology is retained in iPSCs derived from primary cells of FXS patients (FXS-iPSCs) [46,47]. (B) FXS-iPSCs and derived neural cells showed altered gene expression and translation compared to controls [21,52,53,54,55,56,57]. (C) Studies of targets and functions of FMRP showed that there are cell-type-specific [23] and function-specific [58] targets of FMRP [23,58]. (D) Neurodevelopmental abnormalities, including increased NPC proliferation [21,23,53] and altered neurite growth [47,48,53,55,59], were characterized in iPSC-derived models of FXS. (E) FXS-iPSC-derived neural cells exhibited electrophysiological abnormalities [24,59,60,61,62,63]. (F) Pharmacological rescue [64,65,66,67,68,69] and gene editing [70,71,72,73] are two major methods of FMR1 reactivation that have been explored.

2.2. Gene Expression and Translation in the 2D hiPSC-Derived Cell Models of FXS
Following the generation of FXS-iPSCs and confirmation that the molecular features of the disorder are conserved in these cells, several studies have used FXS-iPSCs to reveal altered gene expression in FXS (Figure 1B). A DNA microarray analysis by Halevy et al. (2015) and a more recent study using RNA sequencing by Utami et al. (2020) both showed that several genes associated with neuronal differentiation, neuronal projection, axogenesis, axon guidance, and synaptic signaling were downregulated in neurons derived from FXS-iPSCs compared to controls [52,53]. These genes included ROBO3, SLIT1, DCC, srGAP3, and CRMP. Many downregulated genes in FXS-iPSC-derived neurons were found to bind the repressor element 1 silencing transcription factor (REST), which inhibits neural gene expression in non-neuronal tissues and decreases in expression with the progression of neural differentiation [52,74]. To further elucidate the interaction between FMRP and REST, Halevy et al. (2015) conducted a microRNA (miRNA) array analysis and identified hsa-mir-382, which bound REST and was downregulated in FXS-iPSC-derived neurons. Overexpression of hsa-mir-382 led to an increase in levels of REST target genes involved in axon guidance that were found to be downregulated in FXS-iPSC-derived neurons. This finding indicates that, when FMRP is absent, hsa-mir-382 levels are decreased, leading to decreased repression of REST, which results in a stronger suppression of neural genes essential for normal differentiation and development [52]. Another study using RNA sequencing by Lu et al. (2016) found hundreds of differentially expressed genes (DEGs) in FXS-iPSCs and iPSC-derived neurons compared to control iPSCs and iPSC-derived neurons [54]. Most genes were related to neural development, potassium channels, and glutamatergic synapses. Genes involved in neuronal differentiation and neural development that were upregulated included WNT1, BMP4, POU3F4, TFAP2C, and PAX3, some of which are known to be involved in deterring cells from neuronal differentiation and supporting the maintenance of undifferentiated proliferative states. In contrast, many genes coding for potassium channels, including KCNA1, KCNC3, KCNG2, KCNIP4, KCNJ3, KCNK9, and KCNT1, were downregulated [54]. Boland et al. (2017) confirmed the dysregulated expression of ROBO, SLIT, and another REST target gene, TSPAN7, in neural progenitor cells (NPCs) and neurons derived from FXS-iPSCs [55]. In addition to an altered expression of many genes involved in neuronal development revealed in earlier studies, the authors found differentially expressed genes associated with developmental signaling and cell migration (BAMBI, BMP7, DKK1, and CHCHD2) in FXS-iPSC-derived neural cells compared to controls [55].
In line with findings of altered expression of genes involved in neuronal differentiation [52,54,55] in iPSC models of FXS, several studies have revealed an imbalance in neuronal–glial development. Two studies that used NPCs derived from FMR1-knockout human iPSCs created using the CRISPR/Cas9 system found that the FMR1-null NPCs had elevated expression of glial fibrillary acidic protein (GFAP), a marker for astrocytes [24,56]. Although these cells appeared to have a morphology similar to glial cells, immunofluorescence staining of GFAP-positive cells showed that these cells were positive for SOX2 and NESTIN, both of which are markers for NPCs [56]. The group concluded that these are NPCs with an abnormal morphology and glial gene expression rather than true astrocytes. Interestingly, the elevated GFAP expression was reversible through reintroduction of FMRP into FMR1-null NPCs via a lentiviral vector, and the reduction in GFAP levels was found to be proportional to the increase in expression of FMRP [56].
Recently, a group developed a flow-cytometry-based high-throughput single-cell assay that measures translation and proliferation markers specific to the type of neural cell [21]. Global protein synthesis was elevated in FXS-iPSC-derived NPCs compared to controls (Figure 1B). Moreover, the FXS NPCs appeared to more strongly express markers of proliferation than those of neurogenesis. The translational defects were more obvious in early proliferative cells than more mature cells [21,53]. The group further explored the dysregulated protein synthesis seen in FXS NPCs by investigating the phosphoinositide 3-kinase (PI3K) pathway, which has been found to be overactive in FXS by previous research [75,76,77]. An inhibitor of p110β, a catalytic subunit known to play a key role in driving the hyperactive signaling of the PI3K pathway in FXS, was able to ameliorate the protein synthesis defects in FXS NPCs [21].
A recent study explored a possible mechanism that could explain the dysregulation of genes in FXS. Kurosaki et al. (2021) found that the nonsense-mediated mRNA decay (NMD) pathway modulates the expression of genes via interaction with FMRP [57]. A key NMD factor UPF1 was found to bind directly to FMRP and promote the binding of FMRP to targets of the NMD pathway. FMRP normally acts to suppress NMD, which means NMD is hyperactivated in FXS. Three NMD inhibitors tested in the study (NMDI-1, NMDI-14, and curcumin) were able to partially rescue the expression of neuronal markers and increase neurite growth in FXS-iPSC-derived neurons, although not to the level seen in control iPSC-derived neurons [57]. Of note, however, is that NMD hyperactivation is present in FXS-iPSCs and primary neural cells but absent in neurons derived from these iPSCs, which implies that the process is regulated by other cellular and molecular factors. One example of another mRNA degradation pathway that may play a role in FXS is the N6-methyladenosine (m6A) pathway, which is the most prevalent internal modification mechanism of eukaryotic mRNAs [78,79,80]. One study found that FMRP targets were enriched with m6A markers and FMRP interacts with YTH N6-methyladenosine RNA binding protein 2 (YTHDF2) [79], one of the m6A readers [79,80].
Several studies, as summarized above, have revealed differential gene expression in FXS-iPSCs and iPSC-derived neural cells. Few have investigated potential mechanisms for the altered transcription. Future studies examining the function of FMRP in dysregulated gene expression would further our understanding of the mechanisms of FXS pathology.
2.3. Studies of FMRP Function and Targets
To identify RNA targets bound by FMRP, Li et al. (2020) applied (1) high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP or CLIP-seq) using dorsal and ventral forebrain NPCS and neurons derived from FMRP-tagged hPSCs (both iPSC and ESC lines) and (2) RNA sequencing of the same four cell types derived from isogenic FMR1 knockout hPSC lines [23]. Consistent with other studies of gene expression and transcription of FXS-iPSC-derived neural cells [52,53], the group’s transcriptome analysis revealed downregulated genes associated with neuronal differentiation and synapse formation in FMR1-KO dorsal NPCs. KO NPCs showed a higher level of proliferation and lower level of differentiation than control NPCs [23], as found in other studies previously discussed (Figure 1D) [21,53]. Interestingly, however, there was little overlap between DEGs identified by RNA-seq and FMR1 targets identified by CLIP-seq in each cell type, implying that targets of FMRP are distinct from DEGs in FXS. The authors state that their findings emphasize that FMRP regulates many cellular functions at multiple levels. FMRP targets common to all four types of cells, as well as cell-type-specific targets, were identified. Among the identified type- or location-specific targets of FMRP was PIK3CB in dorsal NPCs and neurons, highlighting the involvement of FMRP in the PI3K pathway [21,75,76,77,81,82]. In ventral neurons, KIF3B, known to be crucial in axon development [83], and CTNNB1, found to modulate ribosome levels, were among the top hits as FMRP targets. Of note, CTNNB1 was found to be downregulated in KO ventral neurons but upregulated in dorsal NPCs, while no difference between KO and controls was seen in other cell types, meaning that the regulation by FMRP is cell-type-specific (Figure 1C). The study also found that, overall, FMRP bound preferentially to longer RNA targets and to coding regions of mRNAs in contrast to 5′ and 3′ UTRs of mice [13]. In NPCs and ventral neurons, a weak preference for mRNAs with G-quadruplex structures was found, in line with several previous studies [84,85,86].
Another recent study examined the role of FMRP on RNA localization by combining techniques of mechanical subcellular fractionation (soma and neurite separation) and RNA sequencing in both FXS-iPSC-derived neurons and FMRP-KO mouse CAD neuronal cells [58]. Among previously identified FMRP targets in three CLIP-seq studies [13,18,87], the authors found a subset of genes that exhibited decreased neurite localization in FXS cell lines and termed them FMRP localization targets. In both human and mouse cell lines, FMRP localization target genes contained more G-quadruplex structures in their 3′ UTRs than genes that were not targets of FMRP. Interestingly, ribosome profiling analysis of FMRP targets in mouse CAD cells showed that this enrichment of G-quadruplexes was absent in FMRP targets of translational regulation, partially explaining the diverging results on FMRP binding motifs in previous research [84,85,86,88]. This finding indicates that there are distinct patterns of FMRP binding to its targets of translational regulation compared to those of subcellular localization (Figure 1C). Moreover, although not tested in hiPSC-derived neurons, the FMRP I304N mutant with dysfunctional translational regulation [89] displayed intact localization of target transcripts in mouse CAD cells, implying two independent functions by FMRP. Further research of FMRP’s functions and targets in humans is necessary.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of relevant findings from primary literature reviewed in text organized by section.
Table 1.
Summary of relevant findings from primary literature reviewed in text organized by section.
| Reference | Section(s) of Review | Model Type(s) | Summary of FXS-iPSC-Relevant Findings |
|---|---|---|---|
| Urbach et al. (2010) [46] | Generation of human iPSC-derived cellular models of FXS
|
|
|
| Sheridan et al. (2011) [47] | Generation of human iPSC-derived cellular models of FXS
|
|
|
| Doers et al. (2014) [48] | Generation of human iPSC-derived cellular models of FXS
|
|
|
| Esanov et al. (2016) [49] | Generation of human iPSC-derived cellular models of FXS
|
|
|
| De Esch et al. (2014) [50] | Generation of human iPSC-derived cellular models of FXS
|
|
|
| Brykczynska et al. (2016) [51] | Generation of human iPSC-derived cellular models of FXS
|
|
|
| Halevy et al. (2015) [52] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS |
|
|
| Utami et al. (2020) [53] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Lu et al. (2016) [54] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS |
|
|
| Boland et al. (2017) [55] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Sunamura et al. (2018) [56] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS |
|
|
| Raj et al. (2021) [21] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS Modeling FXS with human brain organoids |
|
|
| Kurosaki et al. (2021) [57] | Gene expression and translation in the 2D hiPSC-derived cell models of FXS |
|
|
| Li et al. (2020) [23] | Studies of FMRP function and targets |
|
|
| Goering et al. (2020) [58] | Studies of FMRP function and targets |
|
|
| Niedringhaus et al. (2015) [90] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Zhang et al. (2018) [60] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Das Sharma et al. (2020) [61] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Danesi et al. (2018) [63] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Achuta et al. (2017) [62] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS |
|
|
| Achuta et al. (2018) [59] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS
|
|
|
| Brighi et al. (2021) [24] | Characterization of neurodevelopmental and electrophysiological abnormalities of iPSC-derived cell models of FXS
|
|
|
| Bar-Nur et al. (2012) [64] | FMR1 reactivation—Pharmacological rescue |
|
|
| Kaufmann et al. (2015) [65] | FMR1 reactivation—Pharmacological rescue |
|
|
| Kumari et al. (2015) [67] | FMR1 reactivation—Pharmacological rescue |
|
|
| Li et al. (2017) [68] | FMR1 reactivation—Pharmacological rescue |
|
|
| Vershkov et al. (2019) [69] | FMR1 reactivation—Pharmacological rescue |
|
|
| Kumari et al. (2020) [66] | FMR1 reactivation—Pharmacological rescue |
|
|
| Park et al. (2015) [70] | FMR1 reactivation—Gene editing |
|
|
| Xie et al. (2016) [71] | FMR1 reactivation—Gene editing |
|
|
| Liu et al. (2018) [72] | FMR1 reactivation—Gene editing |
|
|
| Graef et al. (2020) [73] | FMR1 reactivation—Gene editing |
|
|
| Kang et al. (2021) [22] | Modeling FXS with human brain organoids |
|
|
FXS = fragile X syndrome, iPSCs = induced pluripotent stem cells, FXS-iPSCs = iPSCs derived from primary cells from individuals with FXS, ESC = embryonic stem cell, NPC = neural progenitor cell, UFM = unmethylated full mutation, MPEP = 2-methyl-6-(phenylethynyl)pyridine, 5-azaC = 5-azacytidine, 5-azadC = 5-aza-2′-deoxycytidine or decitabine, CRISPR = clustered regularly interspaced short palindromic repeats, eCLIP-seq = enhanced crosslinking and immunoprecipitation followed by high-throughput sequencing.
2.4. Characterization of Neurodevelopmental and Electrophysiological Abnormalities of iPSC-Derived Cell Models of FXS
Many studies have focused on the neurodevelopmental features of FXS-iPSCs and iPSC-derived neural cells compared to those of controls. Several groups have found that FXS-iPSC-derived neurons have altered neurite development compared to controls (Figure 1D) [47,48,53,55,59]. Three studies found that FXS neurons have underdeveloped neurites with shorter and fewer neural processes, indicating impairment in both initiating the development of neurites and extending them [47,48,53]. Doers et al. (2014) also reported that axonal growth cones of FXS-iPSC-derived neurons have reduced motility and rates of extension [48]. Another study, through transcriptomic and proteomic analyses, found that genes involved in regulating axonal growth were downregulated in FXS-iPSCs compared to controls [53], which could further explain the undergrowth of neurites. This study also showed that FXS-iPSCs formed significantly smaller neural rosettes than control iPSCs did [53]. However, one study found that immature neurons derived from FXS-iPSCs, in fact, exhibit increased neurite lengths compared to neurons derived from FMR1-expressing iPSCs [55]. The authors state that the diverging results are likely due to differing maturity of the neurons used in the studies. One gene possibly playing a role is SLITRK4, which restrains the growth of neurites, found to be downregulated in NPCs but not in mature neurons [55].
Another group, using a micro-raft array to generate large-scale neuronal cultures, showed that FXS-iPSC-derived neurons have a significant decrease in synaptic vesicle recycling and an increase in unloading of synaptic vesicles compared to control iPSC-derived neurons [90]. These findings were also seen in primary neurons from Fmr1-KO mice compared to those from wildtype mice [90].
Several studies have characterized the electrophysiological properties of FXS-iPSCs and iPSC-derived neural cells (Figure 1E) [24,59,60,61,62,63]. One study found altered homeostatic synaptic plasticity in FXS-iPSC-derived neurons compared to controls [60]. Although FXS neurons did not differ from controls in their baseline amplitude and frequency of miniature excitatory postsynaptic current (mEPSC), nor the inhibitory equivalent (mIPSC), FXS NPCs exhibited impaired retinoic-acid-mediated regulation of synaptic strengths. The increase in mEPSC amplitudes and decrease in mIPSC amplitudes driven by retinoic acid treatment seen in control NPCs were absent in FXS NPCs. Moreover, recovery of FMR1 expression by excision of the expanded CGG repeats using CRISPR/Cas9 normalized the retinoic-acid-mediated synaptic signaling [60].
Whereas the intrinsic properties of mEPSCs or mIPSCs did not appear to differ between FXS and control groups [60,61], FXS-iPSC-derived neurons were found to fire shorter and more frequent spontaneous action potentials than FMR1-expressing controls [61]. The authors then tested the effects of pharmacological treatment on action potential firing in both FXS neurons and controls. While voltage-gated sodium (Na+) channel activator treatment had no significant effect on control neurons, it increased the duration and reduced the frequency of action potentials in FXS neurons, normalizing the firing patterns to resemble those in controls. Furthermore, the treatment of control lines with a persistent Na+ current (Nap) blocker and a calcium (Ca2+)-activated potassium (KCa) channel blocker decreased the duration and increased the frequency of action potential bursts, respectively, both altering the firing patterns to resemble those in FXS neurons [61]. The authors highlight that the study finding of the FXS group displaying a lower conductance of KCa current mediated by the binding of FMRP to the β4 subunit of the KCa channel is consistent with findings in Fmr1-KO mice [91,92]. However, the finding that there is reduced Nap conductance in FXS-iPSC-derived neurons contrasts with findings from mouse models of FXS with increased Nap conductance [93,94], highlighting possible species-specific differences in pathophysiology.
One group that focused on voltage-gated calcium (Cav) channels in FXS-iPSC-derived NPCs and Fmr1 KO mouse NPCs found that the intracellular calcium release signaling in response to membrane depolarization was increased in both human and mouse NPCs lacking FMR1 expression at an early stage of differentiation (day 1) [63]. Moreover, in both humans and mice, the expression of L-type Cav channels was increased in FXS NPCs, indicating the role of this channel type in the enhanced intracellular calcium response [63]. The same group also found that both human FXS NPCs and Fmr1-KO mouse NPCs exhibited larger intracellular calcium release amplitudes in response to the group I mGluR (mGluR1 and mGluR5) agonist (S)-3,5-dihydroxyphenylglycine (DHPG) compared to controls and WTs, respectively [62]. Based on previous research that metabotropic glutamate receptor 5 (mGluR5) signaling is altered in FXS [95,96], the authors investigated the effects of a selective mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) on neuronal differentiation in FXS hiPSC-derived NPCs and Fmr1-KO mouse NPCs [62]. In both human and mouse FXS NPCs, the proportion of cells responsive to DHPG and ionotropic glutamate receptors (iGluRs) was increased compared to controls. Interestingly, however, whereas in human NPCs, MPEP increased the differentiation of NPCs to cells responsive to DHPG and those responsive to both DHPG and iGluRs, MPEP decreased the differentiation of Fmr1-KO mouse NPCs to cells responsive to iGluRs, with no effect on differentiation of DHPG-responsive cells. Furthermore, human FXS NPCs had a larger subpopulation of cells responsive to kainate and NMDA than control NPCs did, but this difference between groups was absent in mouse NPCs. MPEP normalized the increase in NMDA-responsive cells in the FXS group but reduced the kainate-responsive cells in both FXS and control groups. These findings imply that MPEP affects neural development in a species-dependent and cell-type-dependent manner [62] and that glutamatergic signaling pathways may be regulated differently.
Two studies have revealed impairments in maturation of excitatory [24,59] and inhibitory [24] transmission in FXS using iPSC-derived models. One study (Achuta et al., 2018) showed that FXS-iPSC-derived NPCs express a higher level of Ca2+-permeable AMPA receptors (CP-AMPARs) lacking the GluA2 subunit than the controls, representing an aberrant maturation of this subtype of glutamatergic transmission system in FXS [59]. The authors also confirmed this finding in Fmr1-KO mouse NPCs, showing that the process is conserved across species. GluA2-lacking CP-AMPARs are typically found in early developmental stages of neural progenitor differentiation [97,98,99,100]. A larger proportion of FXS NPCs responded to Naspm trihydrochloride (Naspm; a Glu2-lacking CP-AMPAR inhibitor) treatment, also supporting that there are elevated levels of this AMPA receptor type in the FXS group. Based on previous findings that AMPARs can modulate neurite growth [98,99], the authors tested the effect of Naspm on neurite length in iPSC-derived neurons. Naspm reduced the lengths of neurites in both FXS and control neurons so that their lengths were not significantly different from each other, demonstrating that CP-AMPARs have a role in regulating the growth of neural processes [59]. Interestingly, GRIA2, which encodes GluA2, is a target of REST, whose interaction with FMRP is mediated by the miRNA machinery [52]. The authors explain that the lack of Glu2A in FXS NPCs could be due to increased expression of a microRNA miR-181a repressing Glu2A as a consequence of the absence of FMRP [59].
In line with findings by Achuta et al. (2018) of abnormal glutamatergic transmission in FXS [59], Brighi et al. (2021) found that a local application of glutamate triggered a significantly higher increase in intracellular calcium levels in neurons derived from FMR1-knockout human iPSC than in FMRP-expressing neurons [24]. The group also revealed aberrant development of GABA, the main inhibitory neural transmitter, signaling in FMRP-null neurons. Whereas focal GABA application elicited depolarization in about 30% of FMRP-expressing neurons, about 75% of FMRP-null neurons depolarized in response, which indicates that there is a delay in the transition of GABA transmission from excitatory to inhibitory. The GABA and chloride equilibrium reversal has been found to be delayed in Fmr1-KO mice [101] as well.
2.5. Reactivation of FMR1 Gene in FXS
Given that the silencing of FMR1 leads to significant defects in neurodevelopment, many studies have aimed to reactivate FMR1. There are two main approaches, pharmacological agents and gene editing (Figure 1F).
2.5.1. FMR1 Reactivation—Pharmacological Rescue
The most studied pharmacological compounds are the DNA methyltransferase (DNMT) inhibitor 5-azacytidine (5-azaC, azacytidine) and its deoxy derivative 5-aza-2′-deoxycytidine (5-azadC, decitabine) [10,64,65,67,68,69]. In an early study using 5-azaC in FXS-iPSCs and FXS-iPSC-derived neurons, 5-azaC treatment reactivated FMR1 expression to about 15% to 45% of wildtypes (WTs) in FXS-iPSCs [64]. FMR1 expression was restored by 5-azaC in FXS-iPSC-derived neurons as well, but to a lesser degree. The authors describe that 5-azaC can repress DNMT3a and DNMT3b, which are expressed primarily in pluripotent stem cells, possibly explaining the difference in efficacy of 5-azaC in the two cell types. Furthermore, FMR1 reactivation by 5-azaC was retained for at least 7 days after treatment was stopped (not tested past 7 days). 5-azaC treatment substantially reduced the methylation of the FMR1 promoter region in FXS-iPSCs and neurons in a concentration-dependent manner, with the effect being more profound in iPSCs than in neurons. Moreover, following 5-azaC treatment, histone H3 acetylation and H3K4 methylation levels were found to be comparable to those in WT [64].
Two groups independently developed high-throughput drug screening assays to measure FMR1 reactivation in FXS-iPSC-derived neural cells, using high-content imaging [65] or time-resolved fluorescence resonance energy transfer (TR-FRET) [67]. Both studies were to provide proof of principle that the assays are feasible methods for screening possible treatment agents. Although the studies identified a few promising compounds that reactivated FMR1 expression, none were as effective as 5-azaC or 5-azadC without being cytotoxic [65,67].
A few studies have demonstrated methods to further advance high-throughput drug screening. One group developed micro-raft arrays that can be used to expedite screening assays by creating homogenous neuronal cultures at a larger scale. The approach was tested to culture FXS-iPSC-derived neurons and comparing their synaptic features to control iPSC-derived neurons (see Section 2.4 for a summary of findings). Another group created a human iPSC reporter line through a CRISPR/Cas9-based knock-in of a Nano luciferase (Nluc) gene into the human FMR1 gene (FMR1-Nluc reporter line), which can be used to detect FMR1 gene expression with high sensitivity [68].
A small-molecule screening study revealed that FMR1 reactivation in FXS-iPSCs and FXS-iPSC-derived NPCs by 5-azadC was enhanced if used in combination with 3-deazaneplanocin A (DZNep), an inhibitor of both S-adenosyl-homocysteine (SAH) hydrolase and histone methylation [69]. FMR1 reactivation in response to 5-azadC was also verified in vivo in (1) FXS-iPSCs that were injected into immunocompromised mice and (2) FXS-iPSC-derived NPCs transplanted into mouse brains. Moreover, the reactivation was maintained even after 30 days of treatment withdrawal [69]. Another study of small-molecule inhibitors of histone methyl-transferases (HMTs) showed that chaetocin, a fungal toxin that inhibits four main mammalian HMTs (G9a, GLP, SUB39H1, and SETDB1), had a synergistic effect with 5-azadC in reactivating FMR1 in neural stem cells (NSCs) and neurons derived from FXS-iPSCs [66]. These studies highlight the interplay of DNA methylation and histone modifications contributing to FMR1 gene silencing and consequent FXS pathogenesis.
2.5.2. FMR1 Reactivation—Gene Editing
Gene editing approaches to reactivate FMR1 have led to a better understanding of the relationship between CGG repeat expansions and methylation of FMR1, as well as facilitating the development of methods for a more stable restoration of FMR1 expression than by pharmacological agents.
Two groups have shown that reactivation of FMR1 in FXS-iPSCs [70,71] and FXS-iPSC-derived neurons [70] can be achieved by excising out the expanded CGG repeats using CRISPR/Cas9, confirmed by demethylation of the FMR1 promoter, transition to an active chromatin state, and FMRP expression [70,71]. Park et al. (2015) used a double-strand break (DSB) followed by nonhomologous end joining of sequences flanking the CGG repeats [70], whereas Xie et al. (2016) used two DSBs to flank CGG repeats [71]. The former group selected wildtype clones with about 90bp of CGG repeat deletion from their parental lines as the control group, and FXS iPSC clones with similar-sized PCR products to those of the controls as the comparison group. This process resulted in the selection of 2–3 clones out of approximately 100 colonies. On the other hand, the latter group identified five clones with CGG repeat deletions by CRISPR, and only one among the five displayed both FMR1 reactivation and FMRP expression. Although the efficiencies cannot be directly compared between the two studies since the methods and techniques differ, the low overall efficiency of the approach using CRISPR editing to remove CGG repeat expansions warrants further research.
Another group used a DNA methylation editing method by CRISPR/dCas9-Tet1 system to remove the hypermethylation in CGG repeat expansions of the FMR1 locus in FXS-iPSCs and postmitotic neurons derived from FXS-iPSCs [72]. Following targeted demethylation, the FMR1 promoter region transitioned from a heterochromatin state to an active chromatin state, and FMR1 expression was restored in both cell types, although to a lesser extent in postmitotic neurons compared to iPSCs. Furthermore, neurons that were derived from the methylation-edited FXS-iPSCs exhibited normalized electrophysiological phenotypes as examined by a multi-electrode array assay. Persistent expression of FMR1 was detected in vivo in methylation-edited FXS-iPSC-derived neurons transplanted in mouse brains [72].
A recent study measured the threshold of FMR1 reactivation to normalize FXS disease phenotype using a combination of techniques, including CRISPR and antisense oligonucleotides [73]. Results demonstrated that 5% of normal FMRP expression was enough to rescue the elevated spontaneous activity in a mosaic culture of excitatory neurons derived from FXS-iPSCs and FMR1-expressing neurons. Moreover, a neuronal culture in which more than 20% of cells express FMRP had a normal electrophysiological phenotype. However, the reactivation of FMRP was studied in one isogenic line and, thus, verification of results using several different patient cell lines is necessary [73].
As outlined above, both pharmacological and genetic-editing-based reactivation of FMR1 have led to promising results. Partial reactivation of FMR1 may be enough to rescue critical neurodevelopmental abnormalities seen in FXS. Future studies using a standardized protocol for cell cultures and FMR1/FMRP detection assays comparing the efficacies of different genetic editing techniques and their combinations will elucidate the contribution of the different epigenetic mechanisms of FXS pathogenesis. Small molecule screening studies can reveal critical targets interacting with FMRP, which can, in turn, drive genetic editing studies for FMR1 reactivation. Gene editing studies can conversely guide the development of new pharmacological agents specific for FXS.
3. Three-Dimensional (3D) hiPSC-Derived Models of FXS
While 2D models of FXS yielded many remarkable findings of the disease mechanisms, they are somewhat limited as having only one or a few (if co-cultured) cell types for investigation. This does not accurately replicate what is seen in the human brain, where different types of neurons and glial cells co-exist and co-ordinate with one other, forming functional areas that control biological activities in the body [102,103,104]. Therefore, a more sophisticated model system is needed to better recapitulate human neurodevelopment. The development of a recent advanced technique, brain organoids, provides such an opportunity and allows for more accurate modeling of FXS in vitro.
3.1. Brain Organoids—In Vitro System Recapitulating Human Neurodevelopment
Brain organoids are human pluripotent stem cells (hPSC)-derived 3D suspension cultures that recapitulate many features of the developing brain, such as ventricular zone formation, neurogenesis, neuronal migration, transcriptomic and epigenomic profiles, and gliogenesis under long-term culturing [105,106,107,108,109,110,111,112,113]. Brain organoids, therefore, provide a unique platform for investigating normal and abnormal neurodevelopment. The two main categories of brain organoids are (1) cerebral organoids (also known as whole brain organoids), generated via unguided methods that provide no external cues and allow for spontaneous differentiation and self-organization within the organoids [106,112,114,115], and (2) brain-region-specific organoids, generated via protocols guiding the addition of patterning factors (i.e., growth factors and small molecules) to promote the differentiation to a specialized brain region [105,107,110,116,117,118]. Examples of currently available brain-region-specific organoids include forebrain, midbrain, hippocampus, thalamus, hypothalamus, cerebellum, choroid plexus, and ganglionic eminence organoids [105,110,118,119,120,121,122,123,124]. The two types of brain organoids harbor distinct features and advantages/disadvantages. Cerebral organoids, being generated by the spontaneous differentiation capacity of hPSCs, contain a variety of cell lineages, ranging from dorsal forebrain, ventral forebrain, midbrain, hindbrain, and hippocampus, to choroid plexus, retina, astrocytes, and oligodendrocyte [106,112,125,126,127,128,129], whereas brain-region-specific organoids contain only the cell types that are present in the region of interest. Cerebral organoids are, thus, more advantageous in cell lineage diversity, providing opportunities for studying region–region interactions during neurodevelopment. However, the stochastic nature of spontaneous differentiation of hPSCs also brings in additional randomness and heterogeneity to cerebral organoids—i.e., unpredictable and inconsistent proportion and organization of cell lineages within each organoid, making systematic and quantitative analysis difficult and less reliable [130]. From this perspective, brain-region-specific organoids are more advantageous as they contain relatively consistent cell populations with less variability among individuals, improving the reproducibility and reliability of the experiments.
3.2. Modeling FXS with Human Brain Organoids
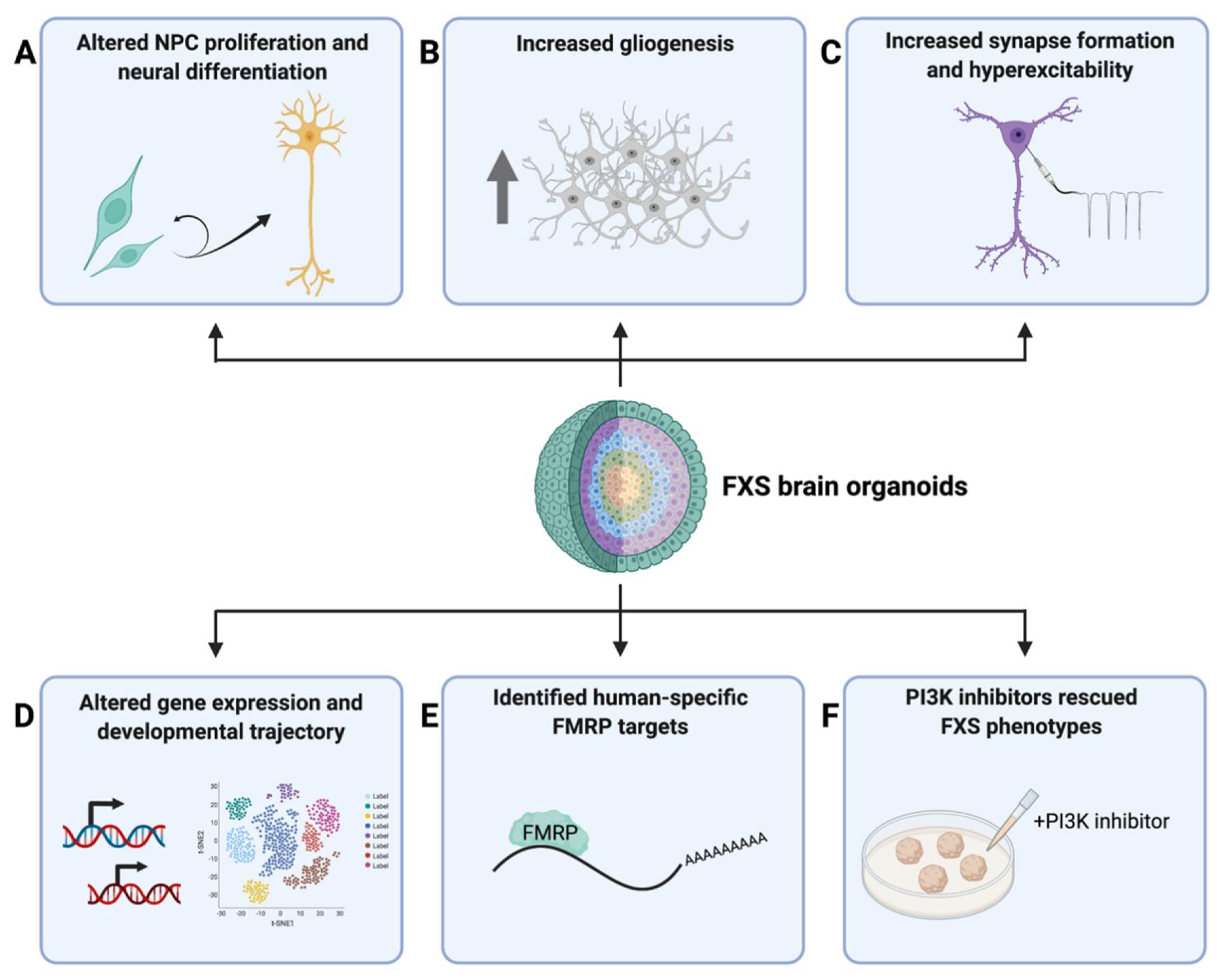
The first study utilizing human brain organoids as a disease model of FXS made use of patient iPSC-derived cortical organoids [21]. Raj et al. (2021) found that FXS cortical organoids at differentiation day 28 exhibited a higher percentage of Ki67+SOX2+ proliferative cells compared to the controls, supporting their findings in 2D cellular models that loss of FMRP led to abnormal proliferation and cell cycle alterations in FXS [21]. Moreover, transcriptomic analysis also revealed a large number of differentially expressed genes (DEGs) between control and FXS cortical organoids. Gene ontology (GO) analysis identified significantly upregulated genes in FXS to be related to proliferation, while significantly downregulated genes in FXS were related to neuronal fate specification, migration, differentiation, and maturation (Figure 2). These results together suggested altered cell-fate decisions in FXS to favor proliferation over differentiation during early neurodevelopment in humans.
FXS cerebral organoids derived from a CRISPR/Cas9-mediated FMRP-KO hiPSC line were investigated in another study [24]. Brighi et al. (2021) found that FMRP-KO organoids not only were bigger in size compared to control organoids, but also had an increased number of glial cells, presumably astrocytes, suggesting an important role of FMRP in regulating gliogenesis and the balance of neural and glial development, as shown with 2D FXS iPSC-derived cellular models (Figure 2) [24,56]. However, the molecular and cellular mechanisms underlying the observed phenotypes were not elucidated.
These questions were investigated in depth in another study published recently [22]. Kang et al. (2021) utilized FXS patient iPSC-derived forebrain organoids for a comprehensive and thorough investigation on how FMRP deficiency affects gene expression, cellular composition, and propagation, as well as neuronal function during human cortical development [22]. FXS forebrain organoids derived from patient iPSC lines were examined at differentiation day 56 (D56), a critical time period that strongly correlates with the mid-fetal period in humans when cortical neurogenesis and FMRP depletion are in play [110,131,132]. By immunostaining with various markers delineating major aspects of cortical development, the authors revealed that the loss of FMRP impaired neurogenesis and cortical development in many ways, including reduced neural progenitor cell (NPC) proliferation, premature neural differentiation, altered cortical layer formation, disrupted differentiation of GABAergic interneurons, as well as accelerated synapse formation that was consistent with the hyperexcitability found by electrophysiological analyses. Notably, as opposed to what was found in Raj et al. (2021), the reduction in NPC proliferation in these D56 FXS organoids could be suggesting a developmental-stage-specific alteration in NPC proliferation rate, which will be an interesting topic to investigate in future studies. At the molecular level, bulk and single-cell RNA-seq revealed pervasive alterations in gene expression profiles and cell-type-specific developmental trajectories in FXS forebrain organoids, which, in combination with other mutant phenotypes, led to the investigation of potential pharmacological approaches that could rescue the phenotypes observed in these FXS organoids. Previous studies on animal models suggested that both genetic and pharmacological inhibitions of mGluR5 receptor were able to rescue both behavioral and synaptic abnormalities caused by the excessive mGluR signaling in FXS [133,134,135]. Additionally, elevated PI3K signaling has been reported in previous FXS models, in which genetic reduction or pharmacological inhibition of PI3K were able to ameliorate disease phenotypes [21,75,81,82]. Therefore, the authors tested the effects of mGluR5 or PI3K inhibition in FXS forebrain organoids. They found that, while mGluR5 antagonists did not rescue mutant phenotypes in these FXS forebrain organoids, PI3K inhibitors rescued NPC proliferation defects and synaptic formation deficits, suggesting potential human-specific disease mechanisms that had not been seen in previous animal models and highlighting a potential therapeutic target for FXS in humans. The authors went on to ask whether there are human-specific FMRP targets in the FXS forebrain organoids and were able to identify a large number of human-specific FMRP targets via eCLIP-seq, one of which is chromodomain helicase DNA-binding protein 2 (CHD2), a well-known risk gene associated with epilepsy, autism spectrum disorder (ASD), and intellectual disability (Figure 2) [136,137]. Together, these studies provide new insights into the human-specific molecular mechanisms of FXS and highlight the potential therapeutic targets for FXS and ASD in general.
4. Limitations and Future Directions
The aforementioned findings using hiPSC-derived cellular models of FXS highlight human-specific molecular mechanisms underlying FXS. However, there are some limitations of the systems. One major limitation of using hiPSC-based cellular models is the difficulty of controlling the many sources of variability. Factors that introduce variability include the diversity in cell lines, differences in reprograming and differentiation protocols, and instability introduced in reprograming [47,48,138]. Variability is greater in brain organoids than in 2D cellular models. The use of multiple cell lines for both control and FXS groups is important to reduce the possibility of making conclusions based on findings specific to selective clones. Consistency in culture and differentiation conditions is also crucial. Detailed documentation of protocols and enabling public access to methods will enhance reproducibility.
3D cellular models are more advantageous in cell type diversity, cellular architecture, and developmental trajectory. However, there are still limitations with the current systems, one of which is the imperfect representation of cellular composition and organization. For example, Kang et al. (2021) reported the percentage of GABAergic inhibitory neurons to be about 10% in their forebrain organoids, which is significantly lower compared to the actual human brain, making investigations of excitation/inhibition balance (E/I balance) and neural network activity difficult. One feasible approach would be to generate fusion organoids, also known as assembloids [139,140,141,142], that have dorsal forebrain and ventral forebrain organoids fused together. Assembloids allow for a closer examination of the interactions between different cell types and brain regions during neurodevelopment. A recent study utilizing cerebral cortex–ganglionic eminence (Cx + GE) assembloids as a disease model of Rett syndrome revealed defects in the balance of excitatory and inhibitory synapses, GE-dependent hypersynchronous neural network activity, and GE-dependent epileptiform changes in Rett syndrome fusion organoids, thus supporting the feasibility of using assembloids in the study of FXS [143].
An underexplored area of research using hiPSC-based models of FXS is investigating the impact of FMRP deficiency on glial development and function. Very few studies have explored hiPSC-derived glial cells in the study of FXS. One group recently published a method to generate astrocytes with forebrain patterning from hPSCs (hASTRO) [144]. Using this method, the group found that the expression of urokinase plasminogen activator (uPA), a protease that regulates the degradation of the extracellular matrix, was increased in FXS hASTRO compared to control hASTRO [145]. Interestingly, the increase in uPA level was correlated with a reduced intracellular calcium response to depolarization in hASTRO [145], which is in contrast to the enhanced intracellular calcium release seen in FXS NPCs [63]. The authors stated that uPA signaling could play a role in modulating synaptic excitation/inhibition balance found to be altered in FXS [146]. The neuronal–glial interaction in FXS needs to be further explored using hiPSC-derived cellular techniques to provide a better understanding of human-specific pathophysiological processes.
In 3D cellular models, only astrocytes and oligodendrocyte progenitor cells have been identified in typical cortical organoids after long-term culturing [109,110,139,147]. While oligodendrocytes function in myelination and metabolic support of neurons, microglia play important roles in brain infections and inflammation. Therefore, the absence of mature oligodendrocytes and microglia could affect neurodevelopment in cortical organoids, leading to the idea of incorporating these essential glial cells into brain organoids for better modeling of neurodevelopment. Several strategies have been tested, including utilizing certain differentiation inducers and promyelinating drugs to promote differentiation and maturation of oligodendrocytes in brain organoids [148,149], as well as co-culturing microglia with brain-region-specific organoids to promote the migration and incorporation of these cells into organoids [150,151,152,153]. However, none of these systems have been used in the study of FXS. Filling this gap would be an intriguing next step.
Finally, how FMRP deficiency affects late-stage neurodevelopment in humans is not well understood. Since most brain organoids only model the early and mid-fetal stage of neurodevelopment [110,111], less is known on late-stage neurodevelopment in FXS. Long-term culturing of brain organoids is a highly challenging task not only because it is time-consuming and labor-intensive, but also because the insufficient delivery of oxygen and nutrients to the inner core of brain organoids hamper the growth of cells and lead to the build-up of a necrotic core inside the organoid [130,154]. Solutions to this issue mainly focus on incorporating a vasculature system into the organoid. Some strategies include co-culturing of brain organoids with endothelial cells [155], induction of endothelial cell differentiation in cerebral organoids [156], overexpression of genes involved in vascular endothelial cell development [157], and transplantation of organoids into an in vivo system (immunodeficient mice) [155]. Other solutions, such as a sliced neocortical organoid system [158], have also been proposed to overcome the diffusion limit in brain organoids. Additionally, the combination of 2D and 3D cellular models is helpful for studying mature cell types in FXS. A study that longitudinally examines 2D and 3D models at multiple developmental stages will be ideal. As we are still at the beginning of understanding FXS in human neurodevelopment, these advanced techniques would serve as essential tools for the investigations along the road.
In summary, future studies of human FXS cellular models should aim to (1) improve reproducibility, (2) enhance representation of neural network structure and organization through the incorporation of glial cell types and use of assembloids, and (3) combine 2D and 3D models to provide a comprehensive examination of neurodevelopment from early to late stages (Figure 3).
Author Contributions
A.L. and J.X. wrote the manuscript. Z.W. and P.J. edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by National Institutes of Health (NS111602 to P.J., HD104458 to P.J. and Z.W., MH123711 to Z.W.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The figures in this review were created with Biorender.com (accessed on 11 March 2022).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rajaratnam, A.; Shergill, J.; Salcedo-Arellano, M.; Saldarriaga, W.; Duan, X.; Hagerman, R. Fragile X Syndrome and fragile X-associated disorders. F1000res 2017, 6, 2112. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Garber, K.B.; Visootsak, J.; Warren, S.T. Fragile X Syndrome. Eur. J. Hum. Genet. 2008, 16, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 219–245. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Ifrim, M.F.; Valdez, A.N.; Raj, N.; Bassell, G.J. Aberrant rna translation in fragile X syndrome: From fmrp mechanisms to emerging therapeutic strategies. Brain Res. 2018, 1693, 24–36. [Google Scholar] [CrossRef]
- Penagarikano, O.; Mulle, J.G.; Warren, S.T. The pathophysiology of fragile X syndrome. Annu. Rev. Genom. Hum. Genet. 2007, 8, 109–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieretti, M.; Zhang, F.; Fu, Y.-H.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of expression of the fmr-1 gene in fragile X syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Verkerk, A.J.M.H.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a gene (Fmr-1) containing a cgg repeat coincident with a breakpoint cluster region exhibiting length Variation In Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Oberlé, I.; Rousseau, F.; Heitz, D.; Kretz, C.; Devys, D.; Hanauer, A.; Boué, J.; Bertheas, M.F.; Mandel, J.L. Instability of a 550-base pair dna segment and abnormal methylation in fragile X syndrome. Science 1991, 252, 1097–1102. [Google Scholar] [CrossRef]
- Coffee, B.; Zhang, F.; Warren, S.T.; Reines, D. Acetylated histones are associated with fmr1 in normal but not fragile X-Syndrome cells. Nat. Genet. 1999, 22, 98–101. [Google Scholar] [CrossRef]
- Hagerman, P.J.; Hagerman, R. Fragile X Syndrome. Curr. Biol. 2021, 31, R273–R275. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Zhao, X. The Molecular biology of fmrp: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. Fmrp stalls ribosomal translocation on mrnas linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Shu, H.; Donnard, E.; Liu, B.; Jung, S.; Wang, R.; Richter, J.D. Fmrp links optimal codons to mrna stability in neurons. Proc. Natl. Acad. Sci. USA 2020, 117, 30400–30411. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.; Sharma, M.R.; Shi, X.; Agrawal, R.K.; Joseph, S. Fragile X mental retardation protein regulates translation by binding directly to the ribosome. Mol. Cell 2014, 54, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Koscielska, K.A.; Cao, Z.; Hulsizer, S.; Grace, N.; Mitchell, G.; Nacey, C.; Githinji, J.; Mcgee, J.; Garcia-Arocena, D.; et al. Signaling Defects in ipsc-derived fragile X premutation neurons. Hum. Mol. Genet. 2012, 21, 3795–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcedo-Arellano, M.J.; Dufour, B.; Mclennan, Y.; Martinez-Cerdeno, V.; Hagerman, R. Fragile X syndrome and associated disorders: Clinical aspects and pathology. Neurobiol. Dis. 2020, 136, 104740. [Google Scholar] [CrossRef]
- Ascano, M., Jr.; Mukherjee, N.; Bandaru, P.; Miller, J.B.; Nusbaum, J.D.; Corcoran, D.L.; Langlois, C.; Munschauer, M.; Dewell, S.; Hafner, M.; et al. Fmrp targets distinct mrna sequence elements to regulate protein expression. Nature 2012, 492, 382–386. [Google Scholar] [CrossRef]
- Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkert, A.J.M.H.; Holden, J.J.A.; Fenwick, R.G.; Warren, S.T.; et al. Variation of the cgg repeat at the fragile x site results in genetic instability: Resolution of the sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular Mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Raj, N.; Mceachin, Z.T.; Harousseau, W.; Zhou, Y.; Zhang, F.; Merritt-Garza, M.E.; Taliaferro, J.M.; Kalinowska, M.; Marro, S.G.; Hales, C.M.; et al. Cell-Type-Specific profiling of human cellular models of fragile X syndrome reveal Pi3k-Dependent defects in translation and neurogenesis. Cell Rep. 2021, 35, 108991. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, Y.; Li, Y.; Han, Y.; Xu, J.; Niu, W.; Li, Z.; Liu, S.; Feng, H.; Huang, W.; et al. A human forebrain organoid model of fragile x syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat. Neurosci. 2021, 24, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Shin, J.; Risgaard, R.D.; Parries, M.J.; Wang, J.; Chasman, D.; Liu, S.; Roy, S.; Bhattacharyya, A.; Zhao, X. Identification of fmr1-regulated molecular networks in human neurodevelopment. Genome Res. 2020, 30, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Brighi, C.; Salaris, F.; Soloperto, A.; Cordella, F.; Ghirga, S.; De Turris, V.; Rosito, M.; Porceddu, P.F.; D’antoni, C.; Reggiani, A.; et al. Novel fragile X Syndrome 2d and 3d brain models based on human isogenic fmrp-ko ipscs. Cell Death Dis. 2021, 12, 498. [Google Scholar] [CrossRef]
- Dahlhaus, R. Of men and mice: Modeling the fragile X syndrome. Front. Mol. Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Bagni, C.; Oostra, B.A. Fragile X syndrome: From protein function to therapy. Am. J. Med. Genet. Part. A 2013, 161, 2809–2821. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models are needed for studying human neurodevelopmental disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, A.; Zhao, X. Human Pluripotent stem cell models of fragile X syndrome. Mol. Cell. Neurosci. 2016, 73, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Abu Diab, M.; Eiges, R. The contribution of pluripotent stem cell (psc)-based models to the study of fragile X syndrome (fxs). Brain Sci. 2019, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, J.; Mientjes, E.; Bakker, C.; Nieuwenhuizen, I.; Severijnen, L.-A.; Van Der Linde, H.; Nelson, D.; Oostra, B.; Willemsen, R. Elevated Fmr1 mrna levels and reduced protein expression in a mouse model with an unmethylated fragile X Full mutation. Exp. Cell Res. 2007, 313, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Entezam, A.; Biacsi, R.; Orrison, B.; Saha, T.; Hoffman, G.E.; Grabczyk, E.; Nussbaum, R.L.; Usdin, K. Regional fmrp deficits and large repeat expansions into the full mutation range in a new fragile x premutation mouse model. Gene 2007, 395, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damdimopoulou, P.; Rodin, S.; Stenfelt, S.; Antonsson, L.; Tryggvason, K.; Hovatta, O. Human embryonic stem cells. Best Pract Res. Clin. Obstet. Gynaecol. 2016, 31, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Eiges, R.; Urbach, A.; Malcov, M.; Frumkin, T.; Schwartz, T.; Amit, A.; Yaron, Y.; Eden, A.; Yanuka, O.; Benvenisty, N.; et al. Developmental study of fragile x syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell 2007, 1, 568–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telias, M.; Segal, M.; Ben-Yosef, D. Neural differentiation of fragile X human embryonic stem cells reveals abnormal patterns of development despite successful neurogenesis. Dev. Biol 2013, 374, 32–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. Fmr1 epigenetic silencing commonly occurs in undifferentiated fragile x-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haenfler, J.M.; Skariah, G.; Rodriguez, C.M.; Monteiro Da Rocha, A.; Parent, J.M.; Smith, G.D.; Todd, P.K. Targeted reactivation of fmr1 transcription in fragile x syndrome embryonic stem cells. Front. Mol. Neurosci. 2018, 11, 282. [Google Scholar] [CrossRef] [Green Version]
- Telias, M.; Kuznitsov-Yanovsky, L.; Segal, M.; Ben-Yosef, D. Functional deficiencies in fragile x neurons derived from human embryonic stem cells. J. Neurosci. 2015, 35, 15295–15306. [Google Scholar] [CrossRef] [Green Version]
- Gerhardt, J.; Tomishima, M.J.; Zaninovic, N.; Colak, D.; Yan, Z.; Zhan, Q.; Rosenwaks, Z.; Jaffrey, S.; Schildkraut, C.L. The dna replication program is altered at the fmr1 locus in fragile x embryonic stem cells. Mol. Cell 2014, 53, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Matthews, K.R.; Morali, D. National human embryo and embryoid research policies: A survey of 22 top research-intensive countries. Regen. Med. 2020, 15, 1905–1917. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Kitambi, S.S.; Chandrasekar, G. Stem cells: A model for screening, discovery and development of drugs. Stem Cells Cloning 2011, 4, 51–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, K.M.; Song, H.; Ming, G.L. Using two- and three-dimensional human ipsc culture systems to model psychiatric disorders. Adv. Neurobiol. 2020, 25, 237–257. [Google Scholar] [CrossRef]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential modeling of fragile x syndrome by human embryonic stem cells and induced-pluripotent stem cells. Cell Stem Cell 2010, 6, 407. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, S.D.; Theriault, K.M.; Reis, S.A.; Zhou, F.; Madison, J.M.; Daheron, L.; Loring, J.F.; Haggarty, S.J. Epigenetic characterization of the fmr1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile x syndrome. PLoS ONE 2011, 6, E26203. [Google Scholar] [CrossRef] [Green Version]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.C.; Abbeduto, L.; Bhattacharyya, A. Ipsc-Derived forebrain neurons from fxs individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef]
- Esanov, R.; Andrade, N.S.; Bennison, S.; Wahlestedt, C.; Zeier, Z. The fmr1 promoter is selectively hydroxymethylated in primary neurons of fragile x syndrome patients. Hum. Mol. Genet. 2016, 25, 4870–4880. [Google Scholar] [CrossRef]
- De Esch, C.E.; Ghazvini, M.; Loos, F.; Schelling-Kazaryan, N.; Widagdo, W.; Munshi, S.T.; Van Der Wal, E.; Douben, H.; Gunhanlar, N.; Kushner, S.A.; et al. Epigenetic characterization of the fmr1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 2014, 3, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Brykczynska, U.; Pecho-Vrieseling, E.; Thiemeyer, A.; Klein, J.; Fruh, I.; Doll, T.; Manneville, C.; Fuchs, S.; Iazeolla, M.; Beibel, M.; et al. Cgg repeat-induced fmr1 silencing depends on the expansion size in human ipscs and neurons carrying unmethylated full mutations. Stem Cell Rep. 2016, 7, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Halevy, T.; Czech, C.; Benvenisty, N. Molecular mechanisms regulating the defects in fragile x syndrome neurons derived from human pluripotent stem cells. Stem Cell Rep. 2015, 4, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utami, K.H.; Skotte, N.H.; Colaço, A.R.; Yusof, N.; Sim, B.; Yeo, X.Y.; Bae, H.G.; Garcia-Miralles, M.; Radulescu, C.I.; Chen, Q.; et al. Integrative analysis identifies key molecular signatures underlying neurodevelopmental deficits in fragile X syndrome. Biol. Psychiatry 2020, 88, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Chen, X.; Feng, Y.; Zeng, Q.; Jiang, C.; Zhu, X.; Fan, G.; Xue, Z. Integrated transcriptome analysis of human ips cells derived from a fragile X syndrome patient during neuronal differentiation. Sci. China Life Sci. 2016, 59, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szucs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef]
- Sunamura, N.; Iwashita, S.; Enomoto, K.; Kadoshima, T.; Isono, F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep. 2018, 8, 11585. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Imamachi, N.; Pröschel, C.; Mitsutomi, S.; Nagao, R.; Akimitsu, N.; Maquat, L.E. Loss of the fragile x syndrome protein fmrp results in misregulation of nonsense-mediated mrna decay. Nat. Cell Biol. 2021, 23, 40–48. [Google Scholar] [CrossRef]
- Goering, R.; Hudish, L.I.; Guzman, B.B.; Raj, N.; Bassell, G.J.; Russ, H.A.; Dominguez, D.; Taliaferro, J.M. Fmrp promotes rna localization to neuronal projections through interact.tions between its rgg domain and G-Quadruplex rna sequences. Elife 2020, 9, E52621. [Google Scholar] [CrossRef]
- Achuta, V.S.; Moykkynen, T.; Peteri, U.K.; Turconi, G.; Rivera, C.; Keinanen, K.; Castren, M.L. Functional changes of ampa responses in human induced pluripotent stem cell-derived neural progenitors in fragile X syndrome. Sci Signal. 2018, 11, eaan8784. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Marro, S.G.; Zhang, Y.; Arendt, K.L.; Patzke, C.; Zhou, B.; Fair, T.; Yang, N.; Sudhof, T.C.; Wernig, M.; et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci. Transl. Med. 2018, 10, 452. [Google Scholar] [CrossRef] [Green Version]
- Das Sharma, S.; Pal, R.; Reddy, B.K.; Selvaraj, B.T.; Raj, N.; Samaga, K.K.; Srinivasan, D.J.; Ornelas, L.; Sareen, D.; Livesey, M.R.; et al. Cortical neurons derived from human pluripotent stem cells lacking fmrp display altered spontaneous firing patterns. Mol. Autism 2020, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Achuta, V.S.; Grym, H.; Putkonen, N.; Louhivuori, V.; Kärkkäinen, V.; Koistinaho, J.; Roybon, L.; Castrén, M.L. Metabotropic glutamate receptor 5 responses dictate differentiation of neural progenitors to nmda-responsive cells in fragile X syndrome. Dev. Neurobiol. 2017, 77, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Danesi, C.; Achuta, V.S.; Corcoran, P.; Peteri, U.-K.; Turconi, G.; Matsui, N.; Albayrak, I.; Rezov, V.; Isaksson, A.; Castrén, M.L. Increased calcium influx through l-type calcium channels in human and mouse neural progenitors lacking fragile X Mental retardation protein. Stem Cell Rep. 2018, 11, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of fmr1 reactivation in fragile-X Induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell. Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-throughput screening using ipsc-derived neuronal progenitors to identify compounds counteracting epigenetic gene silencing in fragile X syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Kumari, D.; Sciascia, N.; Usdin, K. Small molecules targeting h3k9 methylation prevent silencing of reactivated fmr1 alleles in fragile X syndrome patient derived cells. Genes 2020, 11, 356. [Google Scholar] [CrossRef] [Green Version]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-Throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile x syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Ananiev, G.E.; Musser, M.T.; Ness, K.H.; Maglaque, D.L.; Saha, K.; Bhattacharyya, A.; Zhao, X. Establishment of reporter lines for detecting fragile X mental retardation (fmr1) gene reactivation in human neural cells. Stem Cells 2017, 35, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Vershkov, D.; Fainstein, N.; Suissa, S.; Golan-Lev, T.; Ben-Hur, T.; Benvenisty, N. Fmr1 reactivating treatments in fragile X ipsc-derived neural progenitors in vitro and in vivo. Cell Rep. 2019, 26, 2531–2539.e4. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Halevy, T.; Lee, D.R.; Sung, J.J.; Lee, J.S.; Yanuka, O.; Benvenisty, N.; Kim, D.W. Reversion of fmr1 methylation and silencing by editing the triplet repeats in fragile X Ipsc-derived neurons. Cell Rep. 2015, 13, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Xie, N.; Gong, H.; Suhl, J.A.; Chopra, P.; Wang, T.; Warren, S.T. Reactivation of fmr1 by crispr/cas9-mediated deletion of the expanded cgg-repeat of the fragile X chromosome. PLoS ONE 2016, 11, E0165499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of fragile X Syndrome neurons by dna methylation editing of the fmr1 gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graef, J.D.; Wu, H.; Ng, C.; Sun, C.; Villegas, V.; Qadir, D.; Jesseman, K.; Warren, S.T.; Jaenisch, R.; Cacace, A.; et al. Partial fmrp expression is sufficient to normalize neuronal hyperactivity in fragile X neurons. Eur. J. Neurosci. 2020, 51, 2143–2157. [Google Scholar] [CrossRef] [Green Version]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. Rest and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.; Raj, N.; Molinaro, G.; Allen, A.G.; Whyte, A.J.; Gibson, J.R.; Huber, K.M.; Gourley, S.L.; Bassell, G.J. Selective role of the catalytic pi3k subunit p110β in impaired higher order cognition in fragile X syndrome. Cell Rep. 2015, 11, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.; Bassell, G.J. Excess protein synthesis in fxs patient lymphoblastoid cells can be rescued with a p110β-selective inhibitor. Mol. Med. 2012, 18, 336–345. [Google Scholar] [CrossRef]
- Gross, C.; Nakamoto, M.; Yao, X.; Chan, C.-B.; Yim, S.Y.; Ye, K.; Warren, S.T.; Bassell, G.J. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile x syndrome. J. Neurosci. 2010, 30, 10624–10638. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Choe, J.; Park, O.H.; Kim, Y.K. Molecular mechanisms driving mrna degradation by m6a modification. Trends Genet. 2020, 36, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Kang, Y.; Wang, M.; Li, Y.; Xu, T.; Yang, W.; Song, H.; Wu, H.; Shu, Q.; Jin, P. Fragile X mental retardation protein modulates the stability of its m6a-marked messenger rna targets. Hum. Mol. Genet. 2018, 27, 3936–3950. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Lu, A.-Q. The biological function of m6a reader ythdf2 and its role in human disease. Cancer Cell Int. 2021, 21, 109. [Google Scholar] [CrossRef]
- Gross, C.; Banerjee, A.; Tiwari, D.; Longo, F.; White, A.R.; Allen, A.G.; Schroeder-Carter, L.M.; Krzeski, J.C.; Elsayed, N.A.; Puckett, R.; et al. Isoform-selective phosphoinositide 3-kinase inhibition ameliorates a broad range of fragile X syndrome-associated deficits in a mouse model. Neuropsychopharmacology 2019, 44, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.; Chang, C.W.; Kelly, S.M.; Bhattacharya, A.; Mcbride, S.M.; Danielson, S.W.; Jiang, M.Q.; Chan, C.B.; Ye, K.; Gibson, J.R.; et al. Increased expression of the pi3k enhancer pike mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 2015, 11, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichinose, S.; Ogawa, T.; Jiang, X.; Hirokawa, N. The spatiotemporal construction of the axon initial segment via kif3/kap3/trim46 transport under mark2 signaling. Cell Rep. 2019, 28, 2413–2426.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, J.C.; Jensen, K.B.; Jin, P.; Brown, V.; Warren, S.T.; Darnell, R.B. Fragile X mental retardation protein targets g quartet mrnas important for neuronal function. Cell 2001, 107, 489–499. [Google Scholar] [CrossRef]
- Blice-Baum, A.C.; Mihailescu, M.-R. Biophysical characterization of g-quadruplex forming fmr1 mrna and of its interactions with different fragile X mental retardation protein isoforms. RNA 2014, 20, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Suhl, J.A.; Chopra, P.; Anderson, B.R.; Bassell, G.J.; Warren, S.T. Analysis of fmrp mrna target datasets reveals highly associated mrnas mediated by g-quadruplex structures formed via clustered wgga sequences. Hum. Mol. Genet. 2014, 23, 5479–5491. [Google Scholar] [CrossRef] [Green Version]
- Maurin, T.; Lebrigand, K.; Castagnola, S.; Paquet, A.; Jarjat, M.; Popa, A.; Grossi, M.; Rage, F.; Bardoni, B. Hits-Clip in various brain areas reveals new targets and new modalities of rna binding by fragile X mental retardation protein. Nucleic Acids Res. 2018, 46, 6344–6355. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.R.; Chopra, P.; Suhl, J.A.; Warren, S.T.; Bassell, G.J. Identification of consensus binding sites clarifies fmrp binding determinants. Nucleic Acids Res. 2016, 44, 6649–6659. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Absher, D.; Eberhart, D.E.; Brown, V.; Malter, H.E.; Warren, S.T. Fmrp associates with polyribosomes as an mrnp, and the i304n mutation of severe fragile X syndrome abolishes This Association. Mol. Cell 1997, 1, 109–118. [Google Scholar] [CrossRef]
- Niedringhaus, M.; Dumitru, R.; Mabb, A.M.; Wang, Y.; Philpot, B.D.; Allbritton, N.L.; Taylor, A.M. Transferable neuronal mini-cultures to accelerate screening in primary and induced pluripotent stem cell-derived neurons. Sci. Rep. 2015, 5, 8353. [Google Scholar] [CrossRef] [Green Version]
- Deng, P.-Y.; Klyachko, V.A. Increased persistent sodium current causes neuronal hyperexcitability in the entorhinal cortex of fmr1 knockout mice. Cell Rep. 2016, 16, 3157–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routh, B.N.; Rathour, R.K.; Baumgardner, M.E.; Kalmbach, B.E.; Johnston, D.; Brager, D.H. Increased transient na+ conductance and action potential output in layer 2/3 prefrontal cortex neurons of the Fmr1−/Y Mouse. J. Physiol. 2017, 595, 4431–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, P.Y.; Klyachko, V.A. Genetic upregulation of bk channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J. Physiol. 2016, 594, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bonnan, A.; Bony, G.; Ferezou, I.; Pietropaolo, S.; Ginger, M.; Sans, N.; Rossier, J.; Oostra, B.; Lemasson, G. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1−/Y Mice. Nat. Neurosci. 2014, 17, 1701–1709. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role For Metabotropic glutamate receptor 5 (mglur5) in the pathogenesis of fragile X syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Yan, Q.; Rammal, M.; Tranfaglia, M.; Bauchwitz, R. Suppression of two major fragile X syndrome mouse model phenotypes by the mglur5 antagonist mpep. Neuropharmacology 2005, 49, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Jansson, L.C.; Wigren, H.-K.; Nordström, T.; Åkerman, K.E. Functional A-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic acid receptors in differentiating embryonic neural progenitor cells. Neuroreport 2011, 22, 282–287. [Google Scholar] [CrossRef]
- Voss, O.P.; Milne, S.; Sharkey, J.; O’neill, M.J.; Mcculloch, J. Molecular mechanisms of neurite growth with ampa receptor potentiation. Neuropharmacology 2007, 52, 590–597. [Google Scholar] [CrossRef]
- Whitney, N.P.; Peng, H.; Erdmann, N.B.; Tian, C.; Monaghan, D.T.; Zheng, J.C. Calcium-Permeable ampa receptors containing q/r-unedited glur2 direct human neural progenitor cell differentiation to neurons. FASEB J. 2008, 22, 2888–2900. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Nelson, A.D.; Eickstaedt, J.B.; Wallace, K.; Wright, L.S.; Svendsen, C.N. Glutamate enhances proliferation and neurogenesis in human neural progenitor cell cultures derived from the fetal cortex. Eur. J. Neurosci. 2006, 24, 645–653. [Google Scholar] [CrossRef]
- He, Q.; Nomura, T.; Xu, J.; Contractor, A. The developmental switch in gaba polarity is delayed in fragile X mice. J. Neurosci. 2014, 34, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and evolution of the human neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, J.; Dehaene-Lambertz, G.; Kulikova, S.; Poupon, C.; Huppi, P.S.; Hertz-Pannier, L. The early development of brain white matter: A review of imaging studies in fetuses, newborns and infants. Neuroscience 2014, 276, 48–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quezada, S.; Castillo-Melendez, M.; Walker, D.W.; Tolcos, M. Development of the cerebral cortex and the effect of the intrauterine environment. J. Physiol. 2018, 596, 5665–5674. [Google Scholar] [CrossRef]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Goke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Muguruma, K.; Nishiyama, A.; Kawakami, H.; Hashimoto, K.; Sasai, Y. Self-Organization of polarized cerebellar tissue in 3d culture of human pluripotent stem cells. Cell Rep. 2015, 10, 537–550. [Google Scholar] [CrossRef] [Green Version]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3d culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Pasca, S.P. Human astrocyte maturation captured in 3d cerebral cortical spheroids derived from pluripotent stem cells. Neuron 2017, 95, 779–790.e6. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific organoids using mini-bioreactors for modeling zikv exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Lancaster, M.A.; Castanon, R.; Nery, J.R.; Knoblich, J.A.; Ecker, J.R. Cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep. 2016, 17, 3369–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.; Yoon, S.J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-Term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Brauninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-Organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Danjo, T.; Eiraku, M.; Muguruma, K.; Watanabe, K.; Kawada, M.; Yanagawa, Y.; Rubenstein, J.L.; Sasai, Y. Subregional specification of embryonic stem cell-derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J. Neurosci. 2011, 31, 1919–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroof, A.M.; Keros, S.; Tyson, J.A.; Ying, S.W.; Ganat, Y.M.; Merkle, F.T.; Liu, B.; Goulburn, A.; Stanley, E.G.; Elefanty, A.G.; et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013, 12, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, A.; Muguruma, K.; Sasai, Y. Generation of thalamic neurons from mouse embryonic stem cells. Development 2017, 144, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. Foxg1-Dependent dysregulation of gaba/glutamate neuron differentiation in autism spectrum disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-Formation of optic cups and storable stratified neural retina from human escs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, C.R.; Chen, J.; Tang, Y.; Southwell, D.G.; Chalmers, N.; Vogt, D.; Arnold, C.M.; Chen, Y.J.; Stanley, E.G.; Elefanty, A.G.; et al. Functional maturation of hpsc-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 2013, 12, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Pather, S.R.; Huang, W.K.; Zhang, F.; Wong, S.Z.H.; Zhou, H.; Cubitt, B.; Fan, W.; Chen, C.Z.; Xu, M.; et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal sars-cov-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell 2020, 27, 937–950.e9. [Google Scholar] [CrossRef] [PubMed]
- Sloan, S.A.; Andersen, J.; Pasca, A.M.; Birey, F.; Pasca, S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Fleck, J.S.; Sanchis-Calleja, F.; He, Z.; Santel, M.; Boyle, M.J.; Camp, J.G.; Treutlein, B. Resolving organoid brain region identities by mapping single-cell genomic data to reference atlases. Cell Stem Cell 2021, 28, 1148–1159. [Google Scholar] [CrossRef]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchis-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574, 418–422. [Google Scholar] [CrossRef]
- Kanton, S.; Treutlein, B.; Camp, J.G. Single-Cell genomic analysis of human cerebral organoids. Methods Cell Biol. 2020, 159, 229–256. [Google Scholar] [CrossRef]
- Tanaka, Y.; Cakir, B.; Xiang, Y.; Sullivan, G.J.; Park, I.H. Synthetic analyses of single-cell transcriptomes from multiple brain organoids and fetal brain. Cell Rep. 2020, 30, 1682–1689.e3. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Song, H.; Ming, G.L. Brain organoids: Advances, applications and challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [Green Version]
- Agulhon, C.; Blanchet, P.; Kobetz, A.; Marchant, D.; Faucon, N.; Sarda, P.; Moraine, C.; Sittler, A.; Biancalana, V.; Malafosse, A.; et al. Expression of fmr1, fxr1, and fxr2 genes in human prenatal tissues. J. Neuropathol. Exp. Neurol. 1999, 58, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamanini, F.; Willemsen, R.; Van Unen, L.; Bontekoe, C.; Galjaard, H.; Oostra, B.A.; Hoogeveen, A.T. Differential expression of fmr1, fxr1 and fxr2 proteins in human brain and testis. Hum. Mol. Genet. 1997, 6, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bear, M.F.; Huber, K.M.; Warren, S.T. The Mglur Theory Of Fragile X Mental Retardation. Trends Neurosci. 2004, 27, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Dölen, G.; Osterweil, E.; Rao, B.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of fragile x syndrome in mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalon, A.; Sidorov, M.; Ballard, T.M.; Ozmen, L.; Spooren, W.; Wettstein, J.G.; Jaeschke, G.; Bear, M.F.; Lindemann, L. Chronic pharmacological mglu5 inhibition corrects fragile X in adult mice. Neuron 2012, 74, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, T.; Ji, F.; Yuan, Z.; Jiao, J. Chd2 is required for embryonic neurogenesis in the developing cerebral cortex. Stem Cells 2015, 33, 1794–1806. [Google Scholar] [CrossRef]
- Chenier, S.; Yoon, G.; Argiropoulos, B.; Lauzon, J.; Laframboise, R.; Ahn, J.W.; Ogilvie, C.M.; Lionel, A.C.; Marshall, C.R.; Vaags, A.K.; et al. Chd2 haploinsufficiency is associated with developmental delay, intellectual disability, epilepsy and neurobehavioural problems. J. Neurodev. Disord. 2014, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Susco, S.G.; Arias-Garcia, M.A.; Lopez-Huerta, V.G.; Beccard, A.; Bara, A.M.; Moffitt, J.; Korn, J.; Fu, Z.; Barrett, L.E. Fmr1 loss in a human stem cell model reveals early changes to intrinsic membrane excitability. Dev. Biol. 2020, 468, 93–100. [Google Scholar] [CrossRef]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Tanaka, Y.; Cakir, B.; Patterson, B.; Kim, K.Y.; Sun, P.; Kang, Y.J.; Zhong, M.; Liu, X.; Patra, P.; et al. Hesc-Derived thalamic organoids form reciprocal projections when fused with cortical organoids. Cell Stem Cell 2019, 24, 487–497.e7. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of functional human 3d cortico-motor assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Li, M.Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, R.A.; Miranda, O.A.; Buth, J.E.; Mitchell, S.; Ferando, I.; Watanabe, M.; Allison, T.F.; Kurdian, A.; Fotion, N.N.; Gandal, M.J.; et al. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat. Neurosci. 2021, 24, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Peteri, U.-K.; Pitkonen, J.; Utami, K.H.; Paavola, J.; Roybon, L.; Pouladi, M.A.; Castrén, M.L. Generation of the human pluripotent stem-cell-derived astrocyte model with forebrain identity. Brain Sci. 2021, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Peteri, U.-K.; Pitkonen, J.; De Toma, I.; Nieminen, O.; Utami, K.H.; Strandin, T.M.; Corcoran, P.; Roybon, L.; Vaheri, A.; Ethell, I.; et al. Urokinase Plasminogen activator mediates changes in human astrocytes modeling fragile X syndrome. Glia 2021, 69, 2947–2962. [Google Scholar] [CrossRef]
- Gibson, J.R.; Bartley, A.F.; Hays, S.A.; Huber, K.M. Imbalance of neocortical excitation and inhibition and altered up states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 2008, 100, 2615–2626. [Google Scholar] [CrossRef]
- Quadrato, G.; Arlotta, P. Present and future of modeling human brain development in 3d organoids. Curr. Opin. Cell Biol. 2017, 49, 47–52. [Google Scholar] [CrossRef]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pasca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. Ipsc-Derived human microglia-like cells to study neurological diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [Green Version]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muffat, J.; Li, Y.; Omer, A.; Durbin, A.; Bosch, I.; Bakiasi, G.; Richards, E.; Meyer, A.; Gehrke, L.; Jaenisch, R. Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to zika and dengue infections. Proc. Natl. Acad. Sci. USA 2018, 115, 7117–7122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Yuan, X.; Jones, Z.; Vied, C.; Miao, Y.; Marzano, M.; Hua, T.; Sang, Q.A.; Guan, J.; Ma, T.; et al. Functionalization of brain region-specific spheroids with isogenic microglia-like cells. Sci. Rep. 2019, 9, 11055. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef]
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 2020, 26, 766–781.e9. [Google Scholar] [CrossRef]
Figure 2.
Summary of major findings from 3D FXS−iPSC−derived models. (A) FXS-iPSC-derived forebrain organoids exhibited altered NPC proliferation and neural differentiation [21,22]. In particular, increased NPC proliferation was observed at differentiation day 28 [21], while reduced NPC proliferation and accelerated neural differentiation were observed at day 56 [22], suggesting a developmental-stage-specific alteration in NPC proliferation rate. (B) Increased gliogenesis was observed in FMRP-KO cerebral organoids [24]. (C) FXS-iPSC-derived forebrain organoids showed increased synapse formation and hyperexcitability at differentiation day 56 [22]. (D) Transcriptome analysis revealed alterations in gene expression profile and cell-type-specific developmental trajectory in FXS-iPSC-derived forebrain organoids [21,22]. (E) Using human forebrain organoid models, a large number of human-specific FMRP targets were identified via eCLIP-seq [22]. (F) PI3K inhibitors were able to rescue FXS phenotypes in forebrain organoids [22].
Figure 2.
Summary of major findings from 3D FXS−iPSC−derived models. (A) FXS-iPSC-derived forebrain organoids exhibited altered NPC proliferation and neural differentiation [21,22]. In particular, increased NPC proliferation was observed at differentiation day 28 [21], while reduced NPC proliferation and accelerated neural differentiation were observed at day 56 [22], suggesting a developmental-stage-specific alteration in NPC proliferation rate. (B) Increased gliogenesis was observed in FMRP-KO cerebral organoids [24]. (C) FXS-iPSC-derived forebrain organoids showed increased synapse formation and hyperexcitability at differentiation day 56 [22]. (D) Transcriptome analysis revealed alterations in gene expression profile and cell-type-specific developmental trajectory in FXS-iPSC-derived forebrain organoids [21,22]. (E) Using human forebrain organoid models, a large number of human-specific FMRP targets were identified via eCLIP-seq [22]. (F) PI3K inhibitors were able to rescue FXS phenotypes in forebrain organoids [22].

Figure 3.
Summary of future directions for FXS-iPSC research. Future studies of human iPSC-based models of FXS should aim to (A) improve reproducibility, (B) enhance the representation of neural network structure and organization through the incorporation of various glial cell types and use of assembloids, and (C) provide a comprehensive examination of neurodevelopment from early to late stages by developing models that allow for prolonged culture, such as vascularized and sliced organoid models, as well as by corroborating results through a combination of 2D and 3D models.
Figure 3.
Summary of future directions for FXS-iPSC research. Future studies of human iPSC-based models of FXS should aim to (A) improve reproducibility, (B) enhance the representation of neural network structure and organization through the incorporation of various glial cell types and use of assembloids, and (C) provide a comprehensive examination of neurodevelopment from early to late stages by developing models that allow for prolonged culture, such as vascularized and sliced organoid models, as well as by corroborating results through a combination of 2D and 3D models.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lee, A.; Xu, J.; Wen, Z.; Jin, P. Across Dimensions: Developing 2D and 3D Human iPSC-Based Models of Fragile X Syndrome. Cells 2022, 11, 1725. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111725
AMA Style
Lee A, Xu J, Wen Z, Jin P. Across Dimensions: Developing 2D and 3D Human iPSC-Based Models of Fragile X Syndrome. Cells. 2022; 11(11):1725. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111725
Chicago/Turabian StyleLee, Azalea, Jie Xu, Zhexing Wen, and Peng Jin. 2022. "Across Dimensions: Developing 2D and 3D Human iPSC-Based Models of Fragile X Syndrome" Cells 11, no. 11: 1725. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111725
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.