
Unraveling the Pathogenesis of Asthma and Chronic Obstructive Pulmonary Disease Overlap: Focusing on Epigenetic Mechanisms


 , and
, and
Abstract
:1. Introduction
1.1. Asthma and Chronic Obstructive Pulmonary Disease Overlap Is Associated with Higher Frequency and Severity of Exacerbations and Higher Health Care Cost
1.2. Systemic Inflammation in COPD and Asthma Is Driven by Th1 and Th2 Immune Responses, Respectively, while Both Immune Responses May Contribute to Airway Remodeling in ACO
2. Potential Biomarkers for ACO
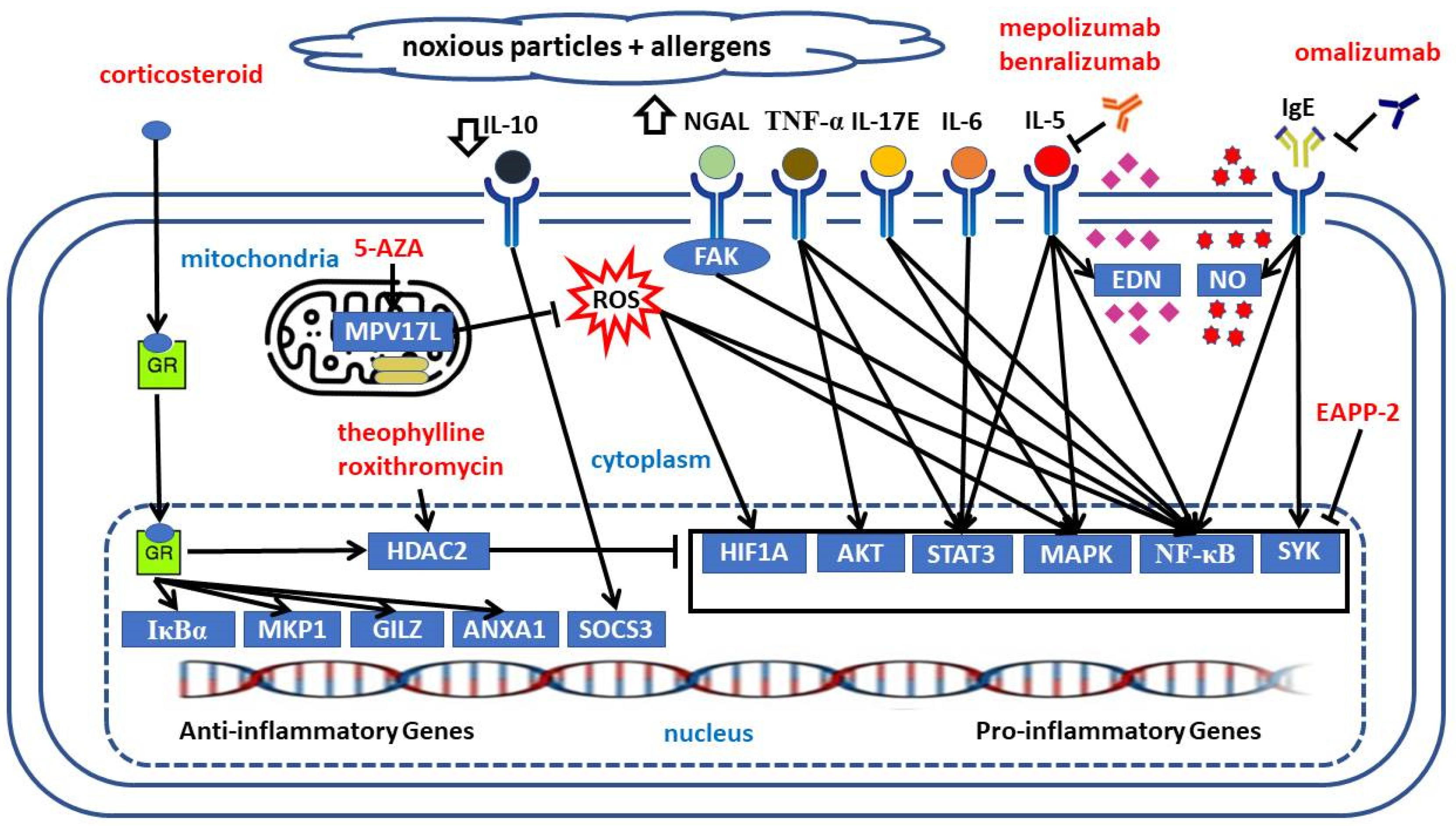
2.1. Neutrophilic Inflammation-Related
2.2. Th2 Response-Related
2.3. Arachidonic Acid-Eicosanoid Pathway-Related
2.4. Metabolites-Related
3. Genetic Variants Associated with ACO
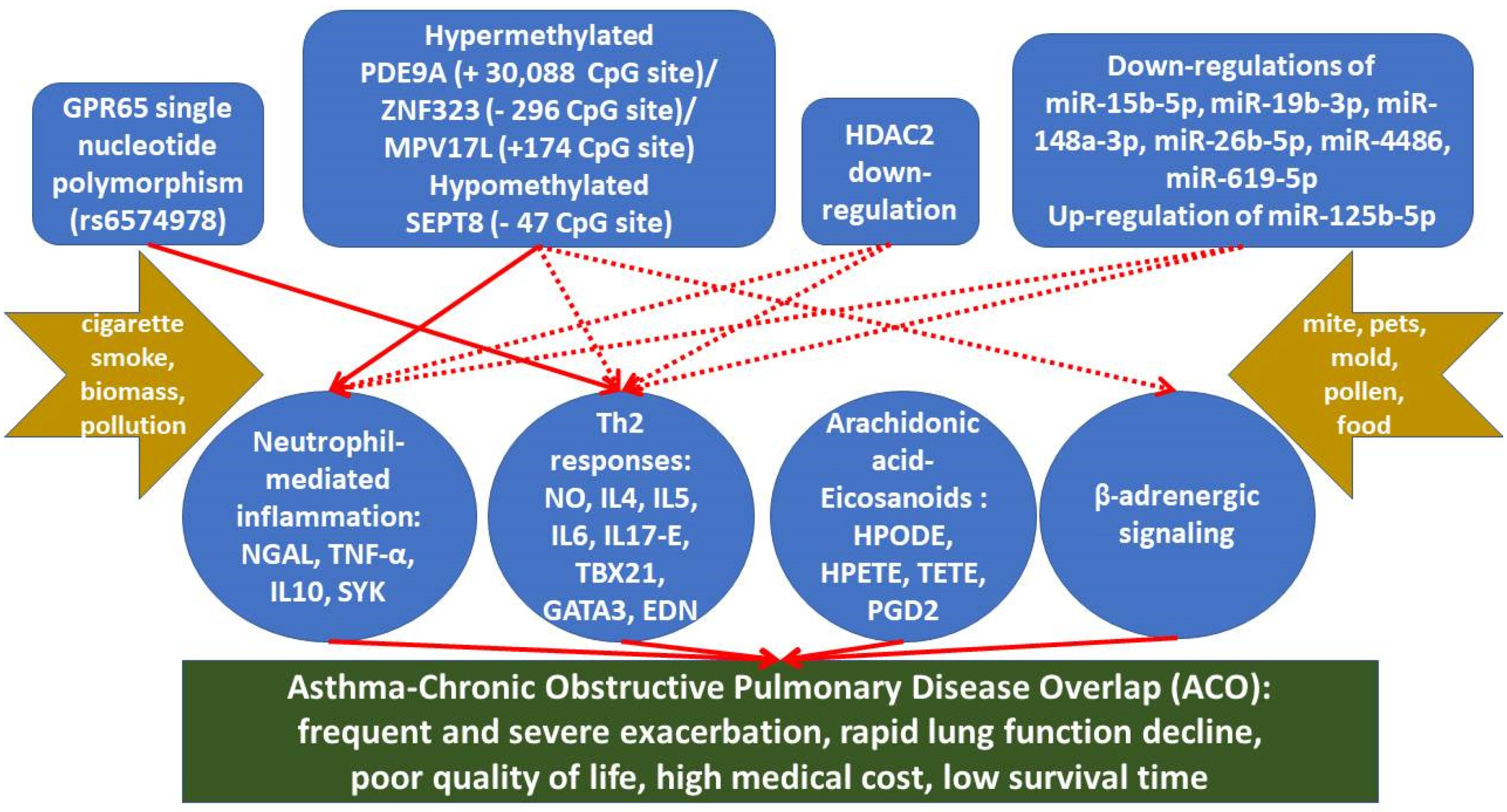
4. Epigenetic Markers Associated with ACO
4.1. Aberrant DNA Methylation
4.2. Histone Modification Patterns and Histone Modifying Enzymes
4.3. MicroRNA Dys-Regulations
5. Limitations and Perspectives of Epigenetic-Related Investigations in ACO
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| COPD | chronic obstructive pulmonary disease |
| ACO | asthma and COPD overlap |
| FEV1 | forced expiratory volume at 1 s |
| FENO | fractional exhaled nitric oxide |
| FVC | forced vital capacity |
| Th | T helper |
| IL | interleukin |
| TNF | tumor necrosis factor |
| NF-κB | nuclear factor kappa B |
| Ig | immunoglobulin |
| NGAL | neutrophil gelatinase-associated lipocalin |
| SYK | spleen associated tyrosine kinase |
| NO | nitric oxide |
| EDN | eosinophil-derived neurotoxin |
| PGD2 | prostaglandin D2 synthase |
| GPR65 | G-protein coupled receptor 65 |
| CpG | cytosine–guanine dinucleotide |
| PDE9A | phosphodiesterase 9A |
| SEPT8 | septin 8 |
| ZNF323/ZSCAN31 | zinc finger and SCAN domain containing 31 |
| MPV17L | MPV17 mitochondrial inner membrane protein like |
| HDAC | histone de-acetylase |
| miRNA | microRNA |
References
- Zhou, X.L.; Zhao, L.Y. Comparison of clinical features and outcomes for asthma-COPD overlap syndrome vs. COPD patients: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1495–1510. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Donovan, C.; Kim, R.Y.; Wark, P.A.B.; Horvat, J.C.; Hansbro, P.M. Asthma-COPD overlap: Current understanding and the utility of experimental models. Eur. Respir. Rev. 2021, 30, 190185. [Google Scholar] [CrossRef]
- Mekov, E.; Nunez, A.; Sin, D.D.; Ichinose, M.; Rhee, C.K.; Maselli, D.J.; Cote, A.; Suppli Ulrik, C.; Maltais, F.; Anzueto, A.; et al. Update on Asthma-COPD Overlap (ACO): A Narrative Review. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Roman-Rodriguez, M.; Kaplan, A. GOLD 2021 Strategy Report: Implications for Asthma-COPD Overlap. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.E.; Wood, J.; Mazurek, J.M. Mortality among Persons with Both Asthma and Chronic Obstructive Pulmonary Disease Aged ≥25 Years, by Industry and Occupation-United States, 1999–2016. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 670–679. [Google Scholar] [CrossRef]
- De Llano, L.P.; Cosío, B.G.; Miravitlles, M.; Plaza, V. Accuracy of a New Algorithm to Identify Asthma-COPD Overlap (ACO) Patients in a Cohort of Patients with Chronic Obstructive Airway Disease. Arch Bronconeumol. 2018, 54, 198–204. [Google Scholar] [CrossRef]
- Park, H.Y.; Lee, S.Y.; Kang, D.; Cho, J.; Lee, H.; Lim, S.Y.; Yoon, H.I.; Ra, S.W.; Kim, K.U.; Oh, Y.M.; et al. Favorable longitudinal change of lung function in patients with asthma-COPD overlap from a COPD cohort. Respir. Res. 2018, 19, 36. [Google Scholar] [CrossRef]
- Mendy, A.; Forno, E.; Niyonsenga, T.; Carnahan, R.; Gasana, J. Prevalence and features of asthma-COPD overlap in the United States 2007–2012. Clin. Respir. J. 2018, 12, 2369–2377. [Google Scholar] [CrossRef]
- Park, S.Y.; Jung, H.; Kim, J.H.; Seo, B.; Kwon, O.Y.; Choi, S.; Oh, B.; Kwon, H.S.; Cho, Y.S.; Moon, H.B.; et al. Longitudinal analysis to better characterize Asthma-COPD overlap syndrome: Findings from an adult asthma cohort in Korea (COREA). Clin. Exp. Allergy 2019, 49, 603–614. [Google Scholar] [CrossRef]
- Karayama, M.; Inui, N.; Yasui, H.; Kono, M.; Hozumi, H.; Suzuki, Y.; Furuhashi, K.; Hashimoto, D.; Enomoto, N.; Fujisawa, T.; et al. Physiological and morphological differences of airways between COPD and asthma-COPD overlap. Sci. Rep. 2019, 9, 7818. [Google Scholar] [CrossRef]
- Gorka, K.; Gross-Sondej, I.; Gorka, J.; Stachura, T.; Polok, K.; Celejewska-Wojcik, N.; Mikrut, S.; Andrychiewicz, A.; Sladek, K.; Soja, J. Assessment of Airway Remodeling Using Endobronchial Ultrasound in Asthma-COPD Overlap. J. Asthma Allergy 2021, 14, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Lin, H.; Huan, C.; Jiang, S.; Lin, D.; Cao, N.; Ren, H. Role of pulmonary function and FeNO detection in early screening of patients with ACO. Exp. Ther. Med. 2020, 20, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Fujino, N.; Sugiura, H. ACO (Asthma-COPD Overlap) Is Independent from COPD, a Case in Favor: A Systematic Review. Diagnostics 2021, 11, 859. [Google Scholar] [CrossRef] [PubMed]
- Sin, D.D.; Miravitlles, M.; Mannino, D.M.; Soriano, J.B.; Price, D.; Celli, B.R.; Leung, J.M.; Nakano, Y.; Park, H.Y.; Wark, P.A.; et al. What is asthma-COPD overlap syndrome? Towards a consensus definition from a round table discussion. Eur. Respir. J. 2016, 48, 664–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo-Parke, H.; Linden, D.; Weldon, S.; Kidney, J.C.; Taggart, C.C. Mechanisms of Virus-Induced Airway Immunity Dysfunction in the Pathogenesis of COPD Disease, Progression, and Exacerbation. Front. Immunol. 2020, 11, 1205. [Google Scholar] [CrossRef]
- Bu, T.; Wang, L.F.; Yin, Y.Q. How Do Innate Immune Cells Contribute to Airway Remodeling in COPD Progression? Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 107–116. [Google Scholar] [CrossRef] [Green Version]
- De Cunto, G.; Cavarra, E.; Bartalesi, B.; Lucattelli, M.; Lungarella, G. Innate Immunity and Cell Surface Receptors in the Pathogenesis of COPD: Insights from Mouse Smoking Models. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 1143–1154. [Google Scholar] [CrossRef]
- Abu Khweek, A.; Kim, E.; Joldrichsen, M.R.; Amer, A.O.; Boyaka, P.N. Insights Into Mucosal Innate Immune Responses in House Dust Mite-Mediated Allergic Asthma. Front. Immunol. 2020, 11, 534501. [Google Scholar] [CrossRef]
- Hikichi, M.; Hashimoto, S.; Gon, Y. Asthma and COPD overlap pathophysiology of ACO. Allergol. Int. 2018, 67, 179–186. [Google Scholar] [CrossRef]
- Toyota, H.; Sugimoto, N.; Kobayashi, K.; Suzuki, Y.; Takeshita, Y.; Ito, A.; Ujino, M.; Tomyo, F.; Sakasegawa, H.; Koizumi, Y.; et al. Comprehensive analysis of allergen-specific IgE in COPD: Mite-specific IgE specifically related to the diagnosis of asthma-COPD overlap. Allergy Asthma Clin. Immunol. 2021, 17, 13. [Google Scholar] [CrossRef]
- Kubysheva, N.; Boldina, M.; Eliseeva, T.; Soodaeva, S.; Klimanov, I.; Khaletskaya, A.; Bayrasheva, V.; Solovyev, V.; Villa-Vargas, L.A.; Ramirez-Salinas, M.A.; et al. Relationship of Serum Levels of IL-17, IL-18, TNF-alpha, and Lung Function Parameters in Patients with COPD, Asthma-COPD Overlap, and Bronchial Asthma. Mediat. Inflamm. 2020, 2020, 4652898. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Choudhury, P.; Kaushik, S.R.; Arya, R.; Nanda, R.; Bhattacharyya, P.; Roychowdhury, S.; Banerjee, R.; Chaudhury, K. Metabolomic fingerprinting and systemic inflammatory profiling of asthma COPD overlap (ACO). Respir. Res. 2020, 21, 126. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Li, S.; Yang, H.; Yuan, J.; Zhang, Z.; Li, X.; Fang, N.; Lin, M.; Hou, Q. Comparative RNA-Seq Transcriptome Analysis on Pulmonary Inflammation in a Mouse Model of Asthma-COPD Overlap Syndrome. Front. Cell Dev. Biol. 2021, 9, 628957. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Good, D.; Mosaiab, T.; Liu, W.; Ni, G.; Kaur, J.; Liu, X.; Jessop, C.; Yang, L.; Fadhil, R.; et al. Significance of LL-37 on Immunomodulation and Disease Outcome. Biomed. Res. Int. 2020, 2020, 8349712. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.S.; Kwon, S.O.; Kim, J.; Kim, W.J. Neutrophil gelatinase-associated lipocalin as a complementary biomarker for the asthma-chronic obstructive pulmonary disease overlap. J. Thorac. Dis. 2018, 10, 5047–5056. [Google Scholar] [CrossRef]
- Yeung, A.C.Y.; Morozov, A.; Robertson, F.P.; Fuller, B.J.; Davidson, B.R. Neutrophil Gelatinase-Associated Lipocalin (NGAL) in predicting acute kidney injury following orthotopic liver transplantation: A systematic review. Int. J. Surg. 2018, 59, 48–54. [Google Scholar] [CrossRef]
- Li, S.; Hui, Y.; Yuan, J.; Zhang, Z.; Li, X.; Fang, N.; Lin, M.; Hou, Q. Syk-Targeted, a New 3-Arylbenzofuran Derivative EAPP-2 Blocks Airway Inflammation of Asthma-COPD Overlap in vivo and in vitro. J. Inflamm. Res. 2021, 14, 2173–2185. [Google Scholar] [CrossRef]
- Ye, M.; Li, Q.; Xiao, L.; Zheng, Z. Serum Magnesium and Fractional Exhaled Nitric Oxide in Relation to the Severity in Asthma-Chronic Obstructive Pulmonary Disease Overlap. Biol. Trace Elem. Res. 2021, 199, 1771–1777. [Google Scholar] [CrossRef]
- Shirai, T.; Hirai, K.; Gon, Y.; Maruoka, S.; Mizumura, K.; Hikichi, M.; Holweg, C.; Itoh, K.; Inoue, H.; Hashimoto, S. Combined Assessment of Serum Periostin and YKL-40 May Identify Asthma-COPD Overlap. J. Allergy Clin. Immunol. 2019, 7, 134–145.e131. [Google Scholar] [CrossRef]
- Hersh, C.P.; Zacharia, S.; Prakash Arivu Chelvan, R.; Hayden, L.P.; Mirtar, A.; Zarei, S.; Putcha, N.; Investigators, C.O. Immunoglobulin E as a Biomarker for the Overlap of Atopic Asthma and Chronic Obstructive Pulmonary Disease. Chronic Obstr. Pulm. Dis. 2020, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yang, T.; He, R.; Li, A.; Dang, W.; Liu, X.; Chen, M. The Value of Inflammatory Biomarkers in Differentiating Asthma-COPD Overlap from COPD. Int. J. Chronic Obstr. Pulm. Dis. 2020, 15, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Uzan, G.C.; Borekci, S.; Doventas, Y.E.; Koldas, M.; Gemicioglu, B. The relationship between inflammatory markers and spirometric parameters in ACOS, Asthma, and COPD. J. Asthma 2020, 57, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Cosio, B.G.; Perez de Llano, L.; Lopez Vina, A.; Torrego, A.; Lopez-Campos, J.L.; Soriano, J.B.; Martinez Moragon, E.; Izquierdo, J.L.; Bobolea, I.; Callejas, J.; et al. Th-2 signature in chronic airway diseases: Towards the extinction of asthma-COPD overlap syndrome? Eur. Respir. J. 2017, 49, 1602397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.Y. GATA3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Shirai, T.; Suzuki, M.; Akamatsu, T.; Suzuki, T.; Hayashi, I.; Yamamoto, A.; Akita, T.; Morita, S.; Asada, K.; et al. A clustering approach to identify and characterize the asthma and chronic obstructive pulmonary disease overlap phenotype. Clin. Exp. Allergy 2017, 47, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Salinas, L.; Verdugo-Rodriguez, A.; Rodriguez, L.L.; Borca, M.V. The Role of Interleukin 6 during Viral Infections. Front. Microbiol. 2019, 10, 1057. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, G.; Umemura, M. Interleukin-17 family cytokines in protective immunity against infections: Role of hematopoietic cell-derived and non-hematopoietic cell-derived interleukin-17s. Microbiol. Immunol. 2018, 62, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, H.F. Eosinophil-Derived Neurotoxin (EDN/RNase 2) and the Mouse Eosinophil-Associated RNases (mEars): Expanding Roles in Promoting Host Defense. Int. J. Mol. Sci. 2015, 16, 15442–15455. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Hirai, K.; Gon, Y.; Maruoka, S.; Mizumura, K.; Hikichi, M.; Itoh, K.; Hashimoto, S. Combined assessment of serum eosinophil-derived neurotoxin and YKL-40 may identify Asthma-COPD overlap. Allergol. Int. 2021, 70, 136–139. [Google Scholar] [CrossRef]
- Cai, C.; Bian, X.; Xue, M.; Liu, X.; Hu, H.; Wang, J.; Zheng, S.G.; Sun, B.; Wu, J.L. Eicosanoids metabolized through LOX distinguish asthma-COPD overlap from COPD by metabolomics study. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1769–1778. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Choudhury, P.; Subramani, E.; Saha, D.; Sengupta, S.; Joshi, M.; Banerjee, R.; Roychowdhury, S.; Bhattacharyya, P.; Chaudhury, K. Metabolomic signatures of asthma-COPD overlap (ACO) are different from asthma and COPD. Metabolomics 2019, 15, 87. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, N.; Choudhury, P.; Joshi, M.; Bhattacharyya, P.; Roychowdhury, S.; Banerjee, R.; Chaudhury, K. Global metabolome profiling of exhaled breath condensates in male smokers with asthma COPD overlap and prediction of the disease. Sci. Rep. 2021, 11, 16664. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Matsuzaki, H.; Mikami, Y.; Makita, K.; Miyakawa, K.; Miyashita, N.; Hosoki, K.; Ishii, T.; Noguchi, S.; Urushiyama, H.; et al. A mouse model of asthma-chronic obstructive pulmonary disease overlap induced by intratracheal papain. Allergy 2021, 76, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, H.; Gao, J.; Koskela, J.; Kinnula, V.; Kobayashi, H.; Laitinen, T.; Mazur, W. Differences in plasma and sputum biomarkers between COPD and COPD-asthma overlap. Eur. Respir. J. 2014, 43, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Tan, X.; Liang, Y.; Hou, C.; Qu, D.; Li, M.; Huang, Q. Differential DAMP release was observed in the sputum of COPD, asthma and asthma-COPD overlap (ACO) patients. Sci. Rep. 2019, 9, 19241. [Google Scholar] [CrossRef] [Green Version]
- Balantic, M.; Rijavec, M.; Flezar, M.; Camlek, T.; Hudoklin, I.; Kosnik, M.; Korosec, P.; Suskovic, S. A polymorphism in ORMDL3 is associated not only with asthma without rhinitis but also with chronic obstructive pulmonary disease. J. Investig. Allergol. Clin. Immunol. 2013, 23, 256–261. [Google Scholar]
- Hayden, L.P.; Cho, M.H.; Raby, B.A.; Beaty, T.H.; Silverman, E.K.; Hersh, C.P.; Investigators, C.O. Childhood asthma is associated with COPD and known asthma variants in COPDGene: A genome-wide association study. Respir. Res. 2018, 19, 209. [Google Scholar] [CrossRef] [Green Version]
- Obeidat, M.; Faiz, A.; Li, X.; van den Berge, M.; Hansel, N.N.; Joubert, P.; Hao, K.; Brandsma, C.A.; Rafaels, N.; Mathias, R.; et al. The pharmacogenomics of inhaled corticosteroids and lung function decline in COPD. Eur. Respir. J. 2019, 54, 1900521. [Google Scholar] [CrossRef]
- Hardin, M.; Cho, M.; McDonald, M.L.; Beaty, T.; Ramsdell, J.; Bhatt, S.; van Beek, E.J.; Make, B.J.; Crapo, J.D.; Silverman, E.K.; et al. The clinical and genetic features of COPD-asthma overlap syndrome. Eur. Respir. J. 2014, 44, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, B.D.; de Jong, K.; Lamontagne, M.; Bosse, Y.; Shrine, N.; Artigas, M.S.; Wain, L.V.; Hall, I.P.; Jackson, V.E.; Wyss, A.B.; et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. 2017, 49, 426–432. [Google Scholar] [CrossRef]
- Sood, A.; Petersen, H.; Blanchette, C.M.; Meek, P.; Picchi, M.A.; Belinsky, S.A.; Tesfaigzi, Y. Methylated Genes in Sputum Among Older Smokers With Asthma. Chest 2012, 142, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.; Zhu, P.; Yang, Z.; Li, M.; Zhang, X.; Cheng, J.; Chen, X.; Lu, F. PCDH20 functions as a tumour-suppressor gene through antagonizing the Wnt/beta-catenin signalling pathway in hepatocellular carcinoma. J. Viral Hepat. 2015, 22, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, C.; Baarsma, H.A.; Wagner, D.E.; Hilgendorff, A.; Konigshoff, M. Linking bronchopulmonary dysplasia to adult chronic lung diseases: Role of WNT signaling. Mol. Cell. Pediatr. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel, L.I.; Almeida, C.B.; Traina, F.; Canalli, A.A.; Dominical, V.M.; Saad, S.T.; Costa, F.F.; Conran, N. Inhibition of phosphodiesterase 9A reduces cytokine-stimulated in vitro adhesion of neutrophils from sickle cell anemia individuals. Inflamm. Res. 2011, 60, 633–642. [Google Scholar] [CrossRef]
- Keele, G.R.; Prokop, J.W.; He, H.; Holl, K.; Littrell, J.; Deal, A.W.; Kim, Y.; Kyle, P.B.; Attipoe, E.; Johnson, A.C.; et al. Sept8/SEPTIN8 involvement in cellular structure and kidney damage is identified by genetic mapping and a novel human tubule hypoxic model. Sci. Rep. 2021, 11, 2071. [Google Scholar] [CrossRef]
- Kurkinen, K.M.; Marttinen, M.; Turner, L.; Natunen, T.; Makinen, P.; Haapalinna, F.; Sarajarvi, T.; Gabbouj, S.; Kurki, M.; Paananen, J.; et al. SEPT8 modulates beta-amyloidogenic processing of APP by affecting the sorting and accumulation of BACE1. J. Cell Sci. 2016, 129, 2224–2238. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Li, X.; Liu, J.; Huo, Y.; Li, L.; Wang, J.; Luo, X.J. Functional variants fine-mapping and gene function characterization provide insights into the role of ZNF323 in schizophrenia pathogenesis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2021, 186, 28–39. [Google Scholar] [CrossRef]
- Chen, Y.C.; Tsai, Y.H.; Wang, C.C.; Liu, S.F.; Chen, T.W.; Fang, W.F.; Lee, C.P.; Hsu, P.Y.; Chao, T.Y.; Wu, C.C.; et al. Epigenome-wide association study on asthma and chronic obstructive pulmonary disease overlap reveals aberrant DNA methylations related to clinical phenotypes. Sci. Rep. 2021, 11, 5022. [Google Scholar] [CrossRef]
- Zwinderman, M.R.H.; de Weerd, S.; Dekker, F.J. Targeting HDAC Complexes in Asthma and COPD. Epigenomes 2019, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Mishra, R.; Chaturvedi, R.; Hashim, Z.; Nath, A.; Khan, A.; Gupta, M.; Singh, H.; Agarwal, V. Role of P-gp and HDAC2 and their Reciprocal Relationship in Uncontrolled Asthma. Curr. Pharm. Biotechnol. 2021, 22, 408–413. [Google Scholar] [CrossRef]
- Hirai, K.; Shirai, T.; Rachi, Y.; Uehara, S.; Ueda, M.; Nakatani, E.; Itoh, K. Impact of Gene Expression Associated with Glucocorticoid-Induced Transcript 1 (GLCCI1) on Severe Asthma and Future Exacerbation. Biol. Pharm. Bull. 2019, 42, 1746–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, M.; Xu, H.; Dai, W.; Zhu, C.; Wu, L.; Yan, S.; Ge, X.; Zhou, W.; Chen, C.; Dai, Y. The role of HDAC2 in cigarette smoke-induced airway inflammation in a murine model of asthma and the effect of intervention with roxithromycin. J. Asthma 2018, 55, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Bersimbaev, R.; Aripova, A.; Bulgakova, O.; Kussainova, C.; Akparova, A.; Izzotti, A. The Plasma Levels of hsa-miR-19b-3p, hsa-miR-125b-5p and hsa-miR-320c in Patients with Asthma, COPD and Asthma-COPD Overlap Syndrome (ACOS). Microrna 2021, 10, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Roffel, M.P.; Maes, T.; Brandsma, C.A.; van den Berge, M.; Vanaudenaerde, B.M.; Joos, G.F.; Brusselle, G.G.; Heijink, I.H.; Bracke, K.R. MiR-223 is increased in lungs of patients with COPD and modulates cigarette smoke-induced pulmonary inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 321, L1091–L1104. [Google Scholar] [CrossRef]
- Xu, W.; Wang, Y.; Ma, Y.; Yang, J. MiR-223 plays a protecting role in neutrophilic asthmatic mice through the inhibition of NLRP3 inflammasome. Respir. Res. 2020, 21, 116. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Feng, Y.; Wu, W.; Chang, C.; Chen, D.; Chen, S.; Zhen, G. microRNA-218-5p plays a protective role in eosinophilic airway inflammation via targeting delta-catenin, a novel catenin in asthma. Clin. Exp. Allergy 2020, 50, 29–40. [Google Scholar] [CrossRef] [PubMed]
- De Smet, E.G.; Van Eeckhoutte, H.P.; Avila Cobos, F.; Blomme, E.; Verhamme, F.M.; Provoost, S.; Verleden, S.E.; Venken, K.; Maes, T.; Joos, G.F.; et al. The role of miR-155 in cigarette smoke-induced pulmonary inflammation and COPD. Mucosal Immunol. 2020, 13, 423–436. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; He, S.; Lu, J.; Liu, C.; Lin, H.; Xu, C.; Xie, L.; Sun, S. MicroRNA-21 aggravates chronic obstructive pulmonary disease by promoting autophagy. Exp. Lung Res. 2018, 44, 89–97. [Google Scholar] [CrossRef]
- Shi, Y.; Fu, X.; Cao, Q.; Mao, Z.; Chen, Y.; Sun, Y.; Liu, Z.; Zhang, Q. Overexpression of miR-155-5p Inhibits the Proliferation and Migration of IL-13-Induced Human Bronchial Smooth Muscle Cells by Suppressing TGF-beta-Activated Kinase 1/MAP3K7-Binding Protein 2. Allergy Asthma Immunol. Res. 2018, 10, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Roffel, M.P.; Bracke, K.R.; Heijink, I.H.; Maes, T. miR-223: A Key Regulator in the Innate Immune Response in Asthma and COPD. Front. Med. 2020, 7, 196. [Google Scholar] [CrossRef]
- Leuenberger, C.; Schuoler, C.; Bye, H.; Mignan, C.; Rechsteiner, T.; Hillinger, S.; Opitz, I.; Marsland, B.; Faiz, A.; Hiemstra, P.S.; et al. MicroRNA-223 controls the expression of histone deacetylase 2: A novel axis in COPD. J. Mol. Med. 2016, 94, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigo-Munoz, J.M.; Rial, M.J.; Sastre, B.; Canas, J.A.; Mahillo-Fernandez, I.; Quirce, S.; Sastre, J.; Cosio, B.G.; Del Pozo, V. Circulating miRNAs as diagnostic tool for discrimination of respiratory disease: Asthma, asthma-chronic obstructive pulmonary disease (COPD) overlap and COPD. Allergy 2019, 74, 2491–2494. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Shirai, T.; Shimoshikiryo, T.; Ueda, M.; Gon, Y.; Maruoka, S.; Itoh, K. Circulating microRNA-15b-5p as a biomarker for asthma-COPD overlap. Allergy 2021, 76, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, M.; Sun, R.; Lin, Z.; Liu, R.; Cai, H.; Tang, Z.; Zhang, R. Methylation of miR-19b-3p promoter exacerbates inflammatory responses in sepsis-induced ALI via targeting KLF7. Cell Biol. Int. 2021, 45, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.M.; Zhang, K.; Zhang, J.H. Human Breast Milk-Derived Exosomal miR-148a-3p Protects Against Necrotizing Enterocolitis by Regulating p53 and Sirtuin 1. Inflammation 2022, 45, 1254–1268. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, Y.; Han, J.; Su, H. GAS5 knockdown ameliorates apoptosis and inflammatory response by modulating miR-26b-5p/Smad1 axis in cerebral ischaemia/reperfusion injury. Behav. Brain Res. 2020, 379, 112370. [Google Scholar] [CrossRef]
- Asensio, V.J.; Tomás, A.; Iglesias, A.; de Llano, L.P.; del Pozo, V.; Cosío, B.G. Eosinophilic COPD Patients Display a Distinctive Serum miRNA Profile from Asthma and Non-eosinophilic COPD. Arch Bronconeumol. 2020, 56, 234–241. [Google Scholar] [CrossRef]
- Roffel, M.P.; Boudewijn, I.M.; van Nijnatten, J.L.L.; Faiz, A.; Vermeulen, C.J.; van Oosterhout, A.J.; Affleck, K.; Timens, W.; Bracke, K.R.; Maes, T.; et al. Identification of asthma associated microRNAs in bronchial biopsies. Eur. Respir. J. 2021, 59, 2101294. [Google Scholar] [CrossRef]
- Yuan, Y.; Xie, S.; Darnell, J.C.; Darnell, A.J.; Saito, Y.; Phatnani, H.; Murphy, E.A.; Zhang, C.; Maniatis, T.; Darnell, R.B. Cell type-specific CLIP reveals that NOVA regulates cytoskeleton interactions in motoneurons. Genome Biol. 2018, 19, 117. [Google Scholar] [CrossRef]
- Krick, S.; Shi, S.; Ju, W.; Faul, C.; Tsai, S.Y.; Mundel, P.; Bottinger, E.P. Mpv17l protects against mitochondrial oxidative stress and apoptosis by activation of Omi/HtrA2 protease. Proc. Natl. Acad. Sci. USA 2008, 105, 14106–14111. [Google Scholar] [CrossRef] [Green Version]
- Amegadzie, J.E.; Gorgui, J.; Acheampong, L.; Gamble, J.M.; Farrell, J.; Gao, Z. Comparative safety and effectiveness of inhaled bronchodilators and corticosteroids for treating asthma-COPD overlap: A systematic review and meta-analysis. J. Asthma 2021, 58, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.S.; Hwang, Y.I.; Yoo, K.H.; Kim, T.H.; Lee, M.G.; Lee, S.H.; Shin, K.C.; In, K.H.; Yoon, H.K.; Rhee, C.K.; et al. Effect of Inhaled Corticosteroids on Exacerbation of Asthma-COPD Overlap According to Different Diagnostic Criteria. J. Allergy Clin. Immunol. Pract 2020, 8, 1625–1633.e1626. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, S.; Kim, J.H.; Kim, S.H.; Lee, T.; Yoon, S.Y.; Kim, M.H.; Moon, J.Y.; Yang, M.S.; Jung, J.W.; et al. A Randomized, Noninferiority Trial Comparing ICS + LABA with ICS + LABA + LAMA in Asthma-COPD Overlap (ACO) Treatment: The ACO Treatment with Optimal Medications (ATOMIC) Study. J. Allergy Clin. Immunol. Pract. 2021, 9, 1304–1311.e1302. [Google Scholar] [CrossRef] [PubMed]
- Maltby, S.; Gibson, P.G.; Powell, H.; McDonald, V.M. Omalizumab Treatment Response in a Population with Severe Allergic Asthma and Overlapping COPD. Chest 2017, 151, 78–89. [Google Scholar] [CrossRef]
- Perez de Llano, L.; Dacal Rivas, D.; Marina Malanda, N.; Plaza Moral, V.; Gullon Blanco, J.A.; Munoz-Esquerre, M.; Garcia-Moguel, I.; Diaz Campos, R.M.; Martinez-Moragon, E.; Harbenau Mena, A.; et al. The Response to Biologics is better in Patients with Severe Asthma Than in Patients with Asthma-COPD Overlap Syndrome. J. Asthma Allergy 2022, 15, 363–369. [Google Scholar] [CrossRef]
- Pavord, I.D.; Chapman, K.R.; Bafadhel, M.; Sciurba, F.C.; Bradford, E.S.; Schweiker Harris, S.; Mayer, B.; Rubin, D.B.; Yancey, S.W.; Paggiaro, P. Mepolizumab for Eosinophil-Associated COPD: Analysis of METREX and METREO. Int. J. Chronic Obstr. Pulm. Dis. 2021, 16, 1755–1770. [Google Scholar] [CrossRef]
- Criner, G.J.; Celli, B.R.; Singh, D.; Agusti, A.; Papi, A.; Jison, M.; Makulova, N.; Shih, V.H.; Brooks, L.; Barker, P.; et al. Predicting response to benralizumab in chronic obstructive pulmonary disease: Analyses of GALATHEA and TERRANOVA studies. Lancet Respir. Med. 2020, 8, 158–170. [Google Scholar] [CrossRef]
- Ranjani, R.; Vinotha, A.T.S. A prospective randomized controlled study: Theophylline on oxidative stress and steroid sensitivity in chronic obstructive pulmonary disease patients. Int. J. Pharm. Investig. 2017, 7, 119–124. [Google Scholar] [CrossRef]
- Li, L.; Pan, Z.Z.; He, J.; Zhou, G.P. Roles of histone acetyltransferase and histone deacetylase in the pathogenesis of bronchial asthma. Zhongguo Dang Dai Er Ke Za Zhi 2015, 17, 629–633. [Google Scholar]
- Tuong, Z.K.; Stewart, B.J.; Guo, S.A.; Clatworthy, M.R. Epigenetics and tissue immunity-Translating environmental cues into functional adaptations. Immunol. Rev. 2022, 305, 111–136. [Google Scholar] [CrossRef]
- Rittiner, J.E.; Moncalvo, M.; Chiba-Falek, O.; Kantor, B. Gene-Editing Technologies Paired With Viral Vectors for Translational Research Into Neurodegenerative Diseases. Front. Mol. Neurosci. 2020, 13, 148. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Expression Levels in ACO | Investigation Model | Main Role | Reference | |
|---|---|---|---|---|
| Neutrophil-mediated inflammation | ||||
| neutrophil gelatinase-associated lipocalin (NGAL) | Increased vs. COPD or asthma | Serum/sputum; ACO rats | Positively correlated with blood eosinophil counts, negatively correlated with FEV1 & FEV1/FVC | [25,39,43,44] |
| TNF-α | Increased vs. COPD or asthma | Serum; ACO mice | [21,22,23] | |
| IL10 | Decreased vs. COPD | serum | [22] | |
| LL37 | Decreased vs. COPD | sputum | Negatively correlated with FEV1, FEV1/FVC, & sputum neutrophil counts | [45] |
| spleen associated tyrosine kinase (SYK) | Increased | ACO mice | Required to form neutrophil extracellular traps; inducing IL-1β, IL-6, and TNF-α via activating NF-κB. | [23,27] |
| Th2 responses | ||||
| nitric oxide | Increased vs. COPD | Exhaled fraction | correlated with FEV1 %predicted | [31] |
| periostin | Increased vs. COPD | serum | Positively correlated with blood eosinophil counts and total IgE | [29] |
| IL4 | Increased vs. COPD or asthma | ACO mice | [23] | |
| IL5 | Increased vs. COPD or asthma | serum | Negatively correlated with FEV1 & FEV1/FVC | [22] |
| IL6 | Increased vs. COPD or asthma | Serum; ACO mice | Negatively correlated with FEV1 & FEV1/FVC | [22,23] |
| IL17-E | Increased vs. COPD | serum | [22] | |
| TBX21/GATA3 | Increased gene expression ratios in ACO vs. COPD or atopic asthma | Peripheral blood mononuclear cells | [35] | |
| eosinophil-derived neurotoxin (EDN) | Increased vs. COPD or asthma | serum | Positively correlated with blood eosinophil counts, negatively with FEV1/FVC | [39] |
| Arachidonic acid-Eicosanoids pathways | ||||
| HPODE | Increased vs. COPD | serum | Negatively correlated with FEV1/FVC | [40] |
| HPETE | Increased vs. COPD | serum | Negatively correlated with FEV1/FVC | [40] |
| HETE | Increased vs. COPD | serum | Negatively correlated with FEV1/FVC | [40] |
| prostaglandin D2 (PGD2) | Increased vs. COPD | serum | Negatively correlated with FEV1/FVC% values | [32] |
| Epigenetic Markers | Changes in ACO | Investigation Model | Potential Mechanisms | Reference |
|---|---|---|---|---|
| PDE9A | Hypermethylated gene body (+30088) in ACO vs. COPD or HS | PBMCs | Augmenting neutrophil adhesion by hydrolysis of cGMP | [54,58] |
| SEPT8 | Hypomethylated gene promoter (−47) in ACO vs. COPD or HS | PBMCs | Augmenting cytokinesis and migration of immune cells | [58,79] |
| ZNF323 | Hypermethylated gene promoter (−296) in ACO vs. COPD or HS | PBMCs | Inhibiting catecholamine synthesis by decreasing tyrosine hydroxylase activity | [57,58] |
| MPV17L | Hypermethylated gene promoter (+174) in ACO vs. healthy subjects | PBMCs; THP1 cell under co-exposure of cigarette smoke extract and ovalbumin | Augmenting mitochondrial oxidative stress and apoptosis | [58,80] |
| HDAC2 | Decreased expression | ACO mice | Desensitizing glucocorticoid receptor | [59] |
| miR-15b-5p | Down-regulated in ACO vs. COPD or asthma | serum | Targeting AKT3, E2F3, MAP2K1, MAPK8, PIK3R1, RAF1, and VEGFA | [72] |
| miR-19b-3p | Down-regulated in ACO vs. COPD | serum | Inhibiting NF-κB signaling via targeting KLF7 | [63,74] |
| miR-125b-5p | Up-regulated in ACO vs. COPD | serum | Promoting NF-κB-mediated inflammation via targeting TNFAIP3 | [63] |
| miR-148a-3p | Down-regulated in ACO vs. COPD or asthma | serum | Inhibiting IKBKB/NF-κB signaling via targeting Tp53 | [72,75] |
| miR-26b-5p | Down-regulated in ACO vs. COPD or asthma | serum | Inhibiting inflammation via targeting SMAD1 | [72,76] |
| miR-4486 | Down-regulated in ACO vs. COPD or asthma | serum | Targeting ERBB2 | [77] |
| miR-619-5p | Down-regulated in ACO vs. COPD or asthma | serum | Targeting ERBB2 | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-C.; Chang, Y.-P.; Huang, K.-T.; Hsu, P.-Y.; Hsiao, C.-C.; Lin, M.-C. Unraveling the Pathogenesis of Asthma and Chronic Obstructive Pulmonary Disease Overlap: Focusing on Epigenetic Mechanisms. Cells 2022, 11, 1728. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111728
Chen Y-C, Chang Y-P, Huang K-T, Hsu P-Y, Hsiao C-C, Lin M-C. Unraveling the Pathogenesis of Asthma and Chronic Obstructive Pulmonary Disease Overlap: Focusing on Epigenetic Mechanisms. Cells. 2022; 11(11):1728. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111728
Chicago/Turabian StyleChen, Yung-Che, Yu-Ping Chang, Kuo-Tung Huang, Po-Yuan Hsu, Chang-Chun Hsiao, and Meng-Chih Lin. 2022. "Unraveling the Pathogenesis of Asthma and Chronic Obstructive Pulmonary Disease Overlap: Focusing on Epigenetic Mechanisms" Cells 11, no. 11: 1728. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11111728