
Biochemical Mechanisms of Sirtuin-Directed Protein Acylation in Hepatic Pathologies of Mitochondrial Dysfunction

Abstract
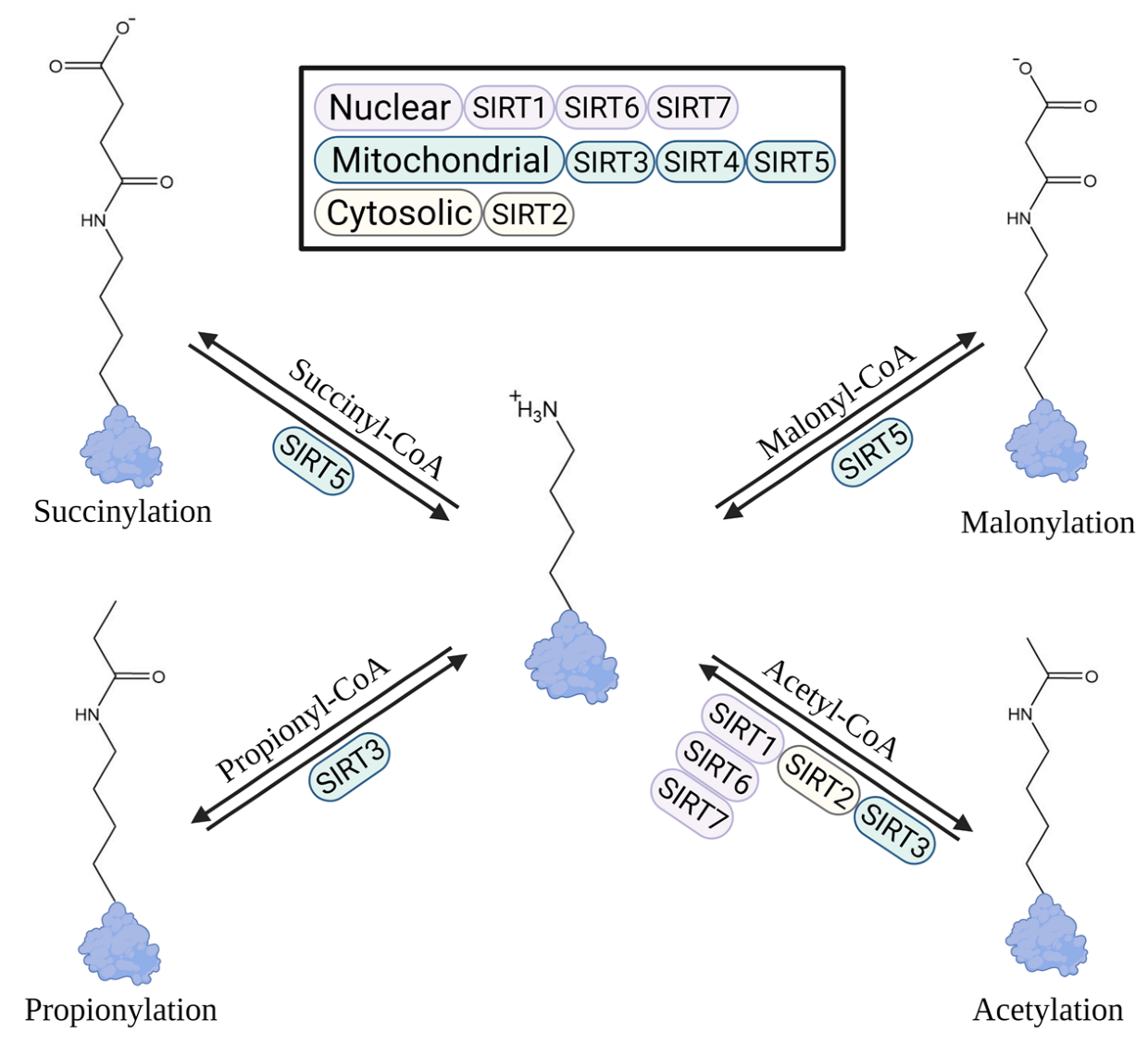
:1. Introduction
2. Biochemical Origins and Dynamics of Lys Acetylation
3. Hepatic Metabolism and Protein Acylation
4. Protein Acetylation and Mitochondrial Dysfunction in Hepatic Pathologies
4.1. Non-Alcoholic Fatty Liver Disease
4.2. Metabolic Syndrome
4.3. Alcohol-Associated Liver Disease
4.4. Hepatocellular Carcinoma
4.5. Aging
5. Conclusions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Seto, W.K.; Mandell, M.S. Chronic liver disease: Global perspectives and future challenges to delivering quality health care. PLoS ONE 2021, 16, e0243607. [Google Scholar] [CrossRef]
- Auger, C.; Alhasawi, A.; Contavadoo, M.; Appanna, V.D. Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front. Cell Dev. Biol. 2015, 3, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezhilarasan, D. Mitochondria: A critical hub for hepatic stellate cells activation during chronic liver diseases. Hepatobiliary Pancreat. Dis. Int. 2021, 20, 315–322. [Google Scholar] [CrossRef]
- Jennings, E.Q.; Fritz, K.S.; Galligan, J.J. Biochemical genesis of enzymatic and non-enzymatic post-translational modifications. Mol. Asp. Med. 2021, 86, 101053. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Meyer, J.G.; Softic, S.; Basisty, N.; Rardin, M.J.; Verdin, E.; Gibson, B.W.; Ilkayeva, O.; Newgard, C.B.; Kahn, C.R.; Schilling, B. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS ONE 2018, 13, e0208973. [Google Scholar] [CrossRef] [Green Version]
- Bak, S.; Leon, I.R.; Jensen, O.N.; Hojlund, K. Tissue specific phosphorylation of mitochondrial proteins isolated from rat liver, heart muscle, and skeletal muscle. J. Proteome Res. 2013, 12, 4327–4339. [Google Scholar] [CrossRef]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef]
- Parodi-Rullan, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [Green Version]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; O’Rourke, B.; Foster, D.B. Metabolism leaves its mark on the powerhouse: Recent progress in post-translational modifications of lysine in mitochondria. Front. Physiol. 2014, 5, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrao, P.; Bork, P.; Krogan, N.J.; van Noort, V. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013, 9, 714. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef]
- Sousa, F.L.; Thiergart, T.; Landan, G.; Nelson-Sathi, S.; Pereira, I.A.; Allen, J.F.; Lane, N.; Martin, W.F. Early bioenergetic evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 368, 20130088. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. Controlled Vocabulary of Posttranslational Modifications (PTM). 2021. Available online: https://www.uniprot.org/docs/ptmlist (accessed on 16 May 2022).
- Wang, Z.A.; Cole, P.A. The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem. Biol. 2020, 27, 953–969. [Google Scholar] [CrossRef]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Gaffney, D.O.; Jennings, E.Q.; Anderson, C.C.; Marentette, J.O.; Shi, T.; Schou Oxvig, A.M.; Streeter, M.D.; Johannsen, M.; Spiegel, D.A.; Chapman, E.; et al. Non-enzymatic Lysine Lactoylation of Glycolytic Enzymes. Cell Chem. Biol. 2020, 27, 206–213.e6. [Google Scholar] [CrossRef]
- James, A.M.; Hoogewijs, K.; Logan, A.; Hall, A.R.; Ding, S.; Fearnley, I.M.; Murphy, M.P. Non-enzymatic N-acetylation of Lysine Residues by AcetylCoA Often Occurs via a Proximal S-acetylated Thiol Intermediate Sensitive to Glyoxalase II. Cell Rep. 2017, 18, 2105–2112. [Google Scholar] [CrossRef] [Green Version]
- Goodman, R.P.; Calvo, S.E.; Mootha, V.K. Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism. J. Biol. Chem. 2018, 293, 7508–7516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trefely, S.; Huber, K.; Liu, J.; Noji, M.; Stransky, S.; Singh, J.; Doan, M.T.; Lovell, C.D.; von Krusenstiern, E.; Jiang, H.; et al. Quantitative subcellular acyl-CoA analysis reveals distinct nuclear metabolism and isoleucine-dependent histone propionylation. Mol. Cell 2022, 82, 447–462.e6. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.P.; Mills, C.A.; Anderson, K.A.; Henriques, B.J.; Lucas, T.G.; Francisco, S.; Liu, J.; Ilkayeva, O.R.; Adams, A.E.; Kulkarni, S.R.; et al. Deglutarylation of glutaryl-CoA dehydrogenase by deacylating enzyme SIRT5 promotes lysine oxidation in mice. J. Biol. Chem. 2022, 298, 101723. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.S.; Gomez, J.D.; Backos, D.S.; Fritz, K.S. Characterizing Sirtuin 3 Deacetylase Affinity for Aldehyde Dehydrogenase 2. Chem. Res. Toxicol. 2017, 30, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, E.Q.; Ray, J.D.; Zerio, C.J.; Trujillo, M.N.; McDonald, D.M.; Chapman, E.; Spiegel, D.A.; Galligan, J.J. Sirtuin 2 Regulates Protein LactoylLys Modifications. Chembiochem 2021, 22, 2102–2106. [Google Scholar] [CrossRef]
- Jin, J.; He, B.; Zhang, X.; Lin, H.; Wang, Y. SIRT2 Reverses 4-Oxononanoyl Lysine Modification on Histones. J. Am. Chem. Soc 2016, 138, 12304–12307. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol. 2004, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Kupis, W.; Palyga, J.; Tomal, E.; Niewiadomska, E. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Neubs, H.P. The nursing shortage: Crisis as opportunity. Fla Nurse 1991, 38, 14. [Google Scholar]
- Long, D.; Wu, H.; Tsang, A.W.; Poole, L.B.; Yoza, B.K.; Wang, X.; Vachharajani, V.; Furdui, C.M.; McCall, C.E. The Oxidative State of Cysteine Thiol 144 Regulates the SIRT6 Glucose Homeostat. Sci. Rep. 2017, 7, 11005. [Google Scholar] [CrossRef] [PubMed]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [Green Version]
- Garrity, J.; Gardner, J.G.; Hawse, W.; Wolberger, C.; Escalante-Semerena, J.C. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J. Biol. Chem. 2007, 282, 30239–30245. [Google Scholar] [CrossRef] [Green Version]
- Tomaselli, D.; Steegborn, C.; Mai, A.; Rotili, D. Sirt4: A Multifaceted Enzyme at the Crossroads of Mitochondrial Metabolism and Cancer. Front. Oncol. 2020, 10, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abril, Y.L.N.; Fernandez, I.R.; Hong, J.Y.; Chiang, Y.L.; Kutateladze, D.A.; Zhao, Q.; Yang, M.; Hu, J.; Sadhukhan, S.; Li, B.; et al. Pharmacological and genetic perturbation establish SIRT5 as a promising target in breast cancer. Oncogene 2021, 40, 1644–1658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tu, B.; Wang, H.; Cao, Z.; Tang, M.; Zhang, C.; Gu, B.; Li, Z.; Wang, L.; Yang, Y.; et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 2014, 111, 10684–10689. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.Y.; Bang, I.H.; Han, C.Y.; Lee, D.H.; Park, B.H.; Bae, E.J. Statin suppresses sirtuin 6 through miR-495, increasing FoxO1-dependent hepatic gluconeogenesis. Theranostics 2020, 10, 11416–11427. [Google Scholar] [CrossRef]
- Rajabi, N.; Galleano, I.; Madsen, A.S.; Olsen, C.A. Targeting Sirtuins: Substrate Specificity and Inhibitor Design. Prog. Mol. Biol. Transl. Sci. 2018, 154, 25–69. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Liang, X.; Roberts, M.S. Hepatic Metabolism in Liver Health and Disease. In Liver Pathophysiology; Academic Press: Cambridge, MA, USA, 2017; pp. 391–400. [Google Scholar]
- Lin, H.; Su, X.; He, B. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 2012, 7, 947–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeva-Andany, M.M.; Perez-Felpete, N.; Fernandez-Fernandez, C.; Donapetry-Garcia, C.; Pazos-Garcia, C. Liver glucose metabolism in humans. Biosci. Rep. 2016, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.G. Hepatic glucose and lipid metabolism. Diabetologia 2016, 59, 1098–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Liu, L.; Gan, W. The Roles of Post-Translational Modifications on mTOR Signaling. Int. J. Mol. Sci. 2021, 22, 1784. [Google Scholar] [CrossRef]
- Assifi, M.M.; Suchankova, G.; Constant, S.; Prentki, M.; Saha, A.K.; Ruderman, N.B. AMP-activated protein kinase and coordination of hepatic fatty acid metabolism of starved/carbohydrate-refed rats. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E794–E800. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Puigserver, P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA 2007, 104, 12861–12866. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [Green Version]
- He, A.; Chen, X.; Tan, M.; Chen, Y.; Lu, D.; Zhang, X.; Dean, J.M.; Razani, B.; Lodhi, I.J. Acetyl-CoA Derived from Hepatic Peroxisomal beta-Oxidation Inhibits Autophagy and Promotes Steatosis via mTORC1 Activation. Mol. Cell 2020, 79, 30–42.e4. [Google Scholar] [CrossRef]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef] [Green Version]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Cox, J.; Weinman, S.A. Forkhead box class O transcription factors in liver function and disease. J. Gastroenterol. Hepatol. 2013, 28, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, B.R.; Scott, I.; Traba, J.; Han, K.; Sack, M.N. Regulation of autophagy and mitophagy by nutrient availability and acetylation. Biochim. Biophys. Acta. 2014, 1841, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Son, S.M.; Park, S.J.; Lee, H.; Siddiqi, F.; Lee, J.E.; Menzies, F.M.; Rubinsztein, D.C. Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 2019, 29, 192–201.e7. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Ozden, O.; Liu, G.; Song, H.Y.; Zhu, Y.; Yan, Y.; Zou, X.; Kang, H.J.; Jiang, H.; Principe, D.R.; et al. SIRT2-Mediated Deacetylation and Tetramerization of Pyruvate Kinase Directs Glycolysis and Tumor Growth. Cancer Res. 2016, 76, 3802–3812. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Xu, Y.P.; Zhao, D.; Li, F.L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.T.; Guan, K.L.; et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell 2013, 52, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Deutsch, R.; Kahen, T.; Lavine, J.E.; Stanley, C.; Behling, C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006, 118, 1388–1393. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003. [Google Scholar] [CrossRef]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Scott, I.; Lu, Z.; Pang, L.; Dimond, C.C.; Gius, D.; Sack, M.N. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med. 2010, 49, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Bharathi, S.S.; Zhang, Y.; Mohsen, A.W.; Uppala, R.; Balasubramani, M.; Schreiber, E.; Uechi, G.; Beck, M.E.; Rardin, M.J.; Vockley, J.; et al. Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site. J. Biol. Chem. 2013, 288, 33837–33847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A.; et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barroso, E.; Rodriguez-Rodriguez, R.; Zarei, M.; Pizarro-Degado, J.; Planavila, A.; Palomer, X.; Villarroya, F.; Vazquez-Carrera, M. SIRT3 deficiency exacerbates fatty liver by attenuating the HIF1alpha-LIPIN 1 pathway and increasing CD36 through Nrf2. Cell Commun. Signal 2020, 18, 147. [Google Scholar] [CrossRef]
- Osborne, B.; Reznick, J.; Wright, L.E.; Sinclair, D.A.C.; Gregory, J. Turner, Nigel Liver-specific overexpression of SIRT3 enhances oxidative metabolism, but does not impact metabolic defects induced by high fat feeding in mice. Biochem. Biophys. Res. Commun. 2022, 607, 131–137. [Google Scholar] [CrossRef]
- Schwer, B.; Bunkenborg, J.; Verdin, R.O.; Andersen, J.S.; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 2006, 103, 10224–10229. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Capra, J.A.; Pollard, K.S.; Verdin, E. SIRT1 and SIRT3 deacetylate homologous substrates: AceCS1,2 and HMGCS1,2. Aging 2011, 3, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxid. 2021, 10, 174. [Google Scholar] [CrossRef]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Hu, B.; Shi, X.; Weidert, E.R.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.E.; Xie, W. Activation of the aryl hydrocarbon receptor sensitizes mice to nonalcoholic steatohepatitis by deactivating mitochondrial sirtuin deacetylase Sirt3. Mol. Cell Biol. 2013, 33, 2047–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, A.; Dean, J.M.; Lu, D.; Chen, Y.; Lodhi, I.J. Hepatic peroxisomal beta-oxidation suppresses lipophagy via RPTOR acetylation and MTOR activation. Autophagy 2020, 16, 1727–1728. [Google Scholar] [CrossRef] [PubMed]
- Bertolio, R.; Napoletano, F.; Mano, M.; Maurer-Stroh, S.; Fantuz, M.; Zannini, A.; Bicciato, S.; Sorrentino, G.; Del Sal, G. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat. Commun. 2019, 10, 1326. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Y.; Cai, C.Z.; Yu, M.L.; Feng, Z.M.; Zhang, Y.W.; Liu, P.H.; Zeng, H.; Yu, C.H. LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 2019, 25, 6607–6618. [Google Scholar] [CrossRef]
- Nassir, F. Role of acetylation in nonalcoholic fatty liver disease: A focus on SIRT1 and SIRT3. Explor. Med. 2020, 1, 248–258. [Google Scholar] [CrossRef]
- Kim, H.; Mendez, R.; Chen, X.; Fang, D.; Zhang, K. Lysine Acetylation of CREBH Regulates Fasting-Induced Hepatic Lipid Metabolism. Mol. Cell Biol. 2015, 35, 4121–4134. [Google Scholar] [CrossRef] [Green Version]
- Bricambert, J.; Miranda, J.; Benhamed, F.; Girard, J.; Postic, C.; Dentin, R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J. Clin. Invest. 2010, 120, 4316–4331. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Feng, B.; George, B.; Chakrabarti, R.; Chen, M.; Chakrabarti, S. Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E127–E137. [Google Scholar] [CrossRef] [Green Version]
- Dall, M.; Hassing, A.S.; Treebak, J.T. NAD(+) and NAFLD caution, causality and careful optimism. J. Physiol. 2021. [Google Scholar] [CrossRef]
- Zhou, C.C.; Yang, X.; Hua, X.; Liu, J.; Fan, M.B.; Li, G.Q.; Song, J.; Xu, T.Y.; Li, Z.Y.; Guan, Y.F.; et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br. J. Pharm. 2016, 173, 2352–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahl, T.B.; Haukeland, J.W.; Yndestad, A.; Ranheim, T.; Gladhaug, I.P.; Damas, J.K.; Haaland, T.; Loberg, E.M.; Arntsen, B.; Birkeland, K.; et al. Intracellular nicotinamide phosphoribosyltransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2010, 95, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Dall, M.; Hassing, A.S.; Niu, L.; Nielsen, T.S.; Ingerslev, L.R.; Sulek, K.; Trammell, S.A.J.; Gillum, M.P.; Barres, R.; Larsen, S.; et al. Hepatocyte-specific perturbation of NAD(+) biosynthetic pathways in mice induces reversible nonalcoholic steatohepatitis-like phenotypes. J. Biol. Chem. 2021, 101388. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Wang, X.N.; Huang, C.C.; Hu, L.; Xiao, Y.F.; Guan, X.H.; Qian, Y.S.; Deng, K.Y.; Xin, H.B. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKalpha/SREBP1 signaling pathway. Lipids Health Dis. 2017, 16, 82. [Google Scholar] [CrossRef]
- Kozako, T.; Suzuki, T.; Yoshimitsu, M.; Arima, N.; Honda, S.; Soeda, S. Anticancer agents targeted to sirtuins. Molecules 2014, 19, 20295–20313. [Google Scholar] [CrossRef] [Green Version]
- Zhao, E.; Hou, J.; Ke, X.; Abbas, M.N.; Kausar, S.; Zhang, L.; Cui, H. The Roles of Sirtuin Family Proteins in Cancer Progression. Cancers 2019, 11, 1949. [Google Scholar] [CrossRef] [Green Version]
- De Lorenzo, S.; Tovoli, F.; Mazzotta, A.; Vasuri, F.; Edeline, J.; Malvi, D.; Boudjema, K.; Renzulli, M.; Jeddou, H.; D’Errico, A.; et al. Non-Alcoholic Steatohepatitis as a Risk Factor for Intrahepatic Cholangiocarcinoma and Its Prognostic Role. Cancers 2020, 12, 3182. [Google Scholar] [CrossRef]
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [Green Version]
- Asgary, S.; Karimi, R.; Momtaz, S.; Naseri, R.; Farzaei, M.H. Effect of resveratrol on metabolic syndrome components: A systematic review and meta-analysis. Rev. Endocr. Metab. Disord. 2019, 20, 173–186. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, X.; Ran, L.; Wan, J.; Wang, X.; Qin, Y.; Shu, F.; Gao, Y.; Yuan, L.; Zhang, Q.; et al. Resveratrol improves insulin resistance, glucose and lipid metabolism in patients with non-alcoholic fatty liver disease: A randomized controlled trial. Dig. Liver Dis. 2015, 47, 226–232. [Google Scholar] [CrossRef]
- Karimi, M.; Abiri, B.; Guest, P.C.; Vafa, M. Therapeutic Effects of Resveratrol on Nonalcoholic Fatty Liver Disease Through Inflammatory, Oxidative Stress, Metabolic, and Epigenetic Modifications. Methods Mol. Biol. 2022, 2343, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yu, X. Efficacy of resveratrol supplementation on liver enzymes in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Complement. Ther. Med. 2021, 57, 102635. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, S.; Mohammadi, H.; Ghavami, A.; Sadeghi, E.; Safari, Z.; Askari, G. Efficacy of resveratrol supplementation in patients with nonalcoholic fatty liver disease: A systematic review and meta-analysis of clinical trials. Complement Clin. Pract. 2021, 42, 101281. [Google Scholar] [CrossRef] [PubMed]
- Seo, D.B.; Jeong, H.W.; Lee, S.J.; Lee, S.J. Coumestrol induces mitochondrial biogenesis by activating Sirt1 in cultured skeletal muscle cells. J. Agric. Food Chem. 2014, 62, 4298–4305. [Google Scholar] [CrossRef]
- Teodoro, J.S.; Duarte, F.V.; Gomes, A.P.; Varela, A.T.; Peixoto, F.M.; Rolo, A.P.; Palmeira, C.M. Berberine reverts hepatic mitochondrial dysfunction in high-fat fed rats: A possible role for SirT3 activation. Mitochondrion 2013, 13, 637–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhu, X.P.; Bai, J.Y.; Xia, P.; Li, Y.; Lu, Y.; Li, X.Y.; Gao, X. Berberine alleviates nonalcoholic fatty liver induced by a high-fat diet in mice by activating SIRT3. FASEB J. 2019, 33, 7289–7300. [Google Scholar] [CrossRef]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxid. Redox Signal 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Huang, L.; Zeng, X.; Li, B.; Wang, C.; Zhou, M.; Lang, H.; Yi, L.; Mi, M. Dihydromyricetin attenuates palmitic acid-induced oxidative stress by promoting autophagy via SIRT3-ATG4B signaling in hepatocytes. Nutr. Metab. 2021, 18, 83. [Google Scholar] [CrossRef]
- Ali, A.; Zhang, Y.; Fu, M.; Pei, Y.; Wu, L.; Wang, R.; Yang, G. Cystathionine gamma-lyase/H2S system suppresses hepatic acetyl-CoA accumulation and nonalcoholic fatty liver disease in mice. Life Sci. 2020, 252, 117661. [Google Scholar] [CrossRef]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef] [Green Version]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, E.J.; Leroith, D.; Karnieli, E. The metabolic syndrome—From insulin resistance to obesity and diabetes. Med. Clin. N. Am. 2011, 95, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.X.; Chaudhary, N.; Akinyemiju, T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988-2012. Prev. Chronic Dis. 2017, 14, E24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bureau, U.S.C. Population Estimates by Age (18+): July 1, 2019. Available online: https://www.census.gov/data/tables/time-series/demo/popest/2010s-national-detail.html (accessed on 2 July 2021).
- Moschen, A.R.; Wieser, V.; Gerner, R.R.; Bichler, A.; Enrich, B.; Moser, P.; Ebenbichler, C.F.; Kaser, S.; Tilg, H. Adipose tissue and liver expression of SIRT1, 3, and 6 increase after extensive weight loss in morbid obesity. J. Hepatol. 2013, 59, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Pieretti, G.; Ciccarelli, F.; Gambardella, A.; Passariello, N.; Rizzo, M.R.; Barbieri, M.; Marfella, R.; Nicoletti, G.; Balestrieri, M.L.; et al. Abdominal Fat SIRT6 Expression and Its Relationship with Inflammatory and Metabolic Pathways in Pre-Diabetic Overweight Patients. Int. J. Mol. Sci. 2019, 20, 1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elhanati, S.; Kanfi, Y.; Varvak, A.; Roichman, A.; Carmel-Gross, I.; Barth, S.; Gibor, G.; Cohen, H.Y. Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 2013, 4, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.; Loureiro, M.P.; Macedo, L.E.; Nocca, D.; Nedelcu, M.; Costa-Casagrande, T.A. Animal models in metabolic syndrome. Rev. Col. Bras. Cir. 2018, 45, e1975. [Google Scholar] [CrossRef] [Green Version]
- Carreira, M.C.; Izquierdo, A.G.; Amil, M.; Casanueva, F.F.; Crujeiras, A.B. Oxidative Stress Induced by Excess of Adiposity Is Related to a Downregulation of Hepatic SIRT6 Expression in Obese Individuals. Oxid. Med. Cell. Longev. 2018, 2018, 6256052. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Botezelli, J.D.; Cambri, L.T.; Ghezzi, A.C.; Dalia, R.A.; Voltarelli, F.A.; de Mello, M.A. Fructose-rich diet leads to reduced aerobic capacity and to liver injury in rats. Lipids Health Dis. 2012, 11, 78. [Google Scholar] [CrossRef] [Green Version]
- Sato, N. Central role of mitochondria in metabolic regulation of liver pathophysiology. J. Gastroenterol. Hepatol. 2007, 22 Suppl. 1, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Mitsuya, M.; Nishimura, T.; Eiki, J.; Nagata, Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure 2004, 12, 429–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, M.A.; Birnbaum, M.J. Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. 2021, 33, 2329–2354. [Google Scholar] [CrossRef] [PubMed]
- Chyau, C.C.; Wang, H.F.; Zhang, W.J.; Chen, C.C.; Huang, S.H.; Chang, C.C.; Peng, R.Y. Antrodan Alleviates High-Fat and High-Fructose Diet-Induced Fatty Liver Disease in C57BL/6 Mice Model via AMPK/Sirt1/SREBP-1c/PPARgamma Pathway. Int. J. Mol. Sci. 2020, 21, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef]
- Pfluger, P.T.; Herranz, D.; Velasco-Miguel, S.; Serrano, M.; Tschop, M.H. Sirt1 protects against high-fat diet-induced metabolic damage. Proc. Natl. Acad. Sci. USA 2008, 105, 9793–9798. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- de Ligt, M.; Bergman, M.; Fuentes, R.M.; Essers, H.; Moonen-Kornips, E.; Havekes, B.; Schrauwen-Hinderling, V.B.; Schrauwen, P. No effect of resveratrol supplementation after 6 months on insulin sensitivity in overweight adults: A randomized trial. Am. J. Clin. Nutr. 2020, 112, 1029–1038. [Google Scholar] [CrossRef]
- Kjaer, T.N.; Ornstrup, M.J.; Poulsen, M.M.; Stodkilde-Jorgensen, H.; Jessen, N.; Jorgensen, J.O.L.; Richelsen, B.; Pedersen, S.B. No Beneficial Effects of Resveratrol on the Metabolic Syndrome: A Randomized Placebo-Controlled Clinical Trial. J. Clin. Endocrinol. Metab. 2017, 102, 1642–1651. [Google Scholar] [CrossRef] [Green Version]
- Goyal, S.N.; Reddy, N.M.; Patil, K.R.; Nakhate, K.T.; Ojha, S.; Patil, C.R.; Agrawal, Y.O. Challenges and issues with streptozotocin-induced diabetes A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Biol. Interact. 2016, 244, 49–63. [Google Scholar] [CrossRef]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoogian, E.N.; Chow, L.S.; Taub, P.R.; Laferrere, B.; Panda, S. Time-restricted eating for the prevention and management of metabolic diseases. Endocr. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Swiatkiewicz, I.; Mila-Kierzenkowska, C.; Wozniak, A.; Szewczyk-Golec, K.; Nuszkiewicz, J.; Wroblewska, J.; Rajewski, P.; Eussen, S.; Faerch, K.; Manoogian, E.N.C.; et al. Pilot Clinical Trial of Time-Restricted Eating in Patients with Metabolic Syndrome. Nutrients 2021, 13, 346. [Google Scholar] [CrossRef]
- Dollerup, O.L.; Christensen, B.; Svart, M.; Schmidt, M.S.; Sulek, K.; Ringgaard, S.; Stodkilde-Jorgensen, H.; Moller, N.; Brenner, C.; Treebak, J.T.; et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 2018, 108, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Kady, R.R.; Ali, A.K.; El Wakeel, L.M.; Sabri, N.A.; Shawki, M.A. Nicotinamide supplementation in diabetic nonalcoholic fatty liver disease patients: Randomized controlled trial. Adv. Chronic. Dis. 2022, 13, 20406223221077958. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Yoshino, J.; Kayser, B.D.; Patti, G.J.; Franczyk, M.P.; Mills, K.F.; Sindelar, M.; Pietka, T.; Patterson, B.W.; Imai, S.I.; et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 2021, 372, 1224–1229. [Google Scholar] [CrossRef]
- Kleckner, A.; Reschke, J.E.; Altman, B.J.; Belcher, E.; Dunne, R.F.; Fleming, F.J.; Gilmore, N.; Jensen-Battaglia, M.; Kleckner, I.; Lin, P.-J.; et al. A 10-hour time-restricted eating intervention to address cancer-related fatigue among cancer survivors. J. Clin. Oncol. 2021, 39, 12109. [Google Scholar] [CrossRef]
- Niu, K.M.; Bao, T.; Gao, L.; Ru, M.; Li, Y.; Jiang, L.; Ye, C.; Wang, S.; Wu, X. The Impacts of Short-Term NMN Supplementation on Serum Metabolism, Fecal Microbiota, and Telomere Length in Pre-Aging Phase. Front. Nutr. 2021, 8, 756243. [Google Scholar] [CrossRef]
- Tomiyama, A.J.; Milush, J.M.; Lin, J.; Flynn, J.M.; Kapahi, P.; Verdin, E.; Sinclair, E.; Melov, S.; Epel, E.S. Long-term calorie restriction in humans is not associated with indices of delayed immunologic aging: A descriptive study. Nutr. Healthy Aging 2017, 4, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Wilson, D.F.; Matschinsky, F.M. Ethanol metabolism: The good, the bad, and the ugly. Med. Hypotheses 2020, 140, 109638. [Google Scholar] [CrossRef] [PubMed]
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown—Focus on adducts. Alcohol. Res. Health 2003, 27, 285–290. [Google Scholar] [PubMed]
- Fritz, K.S.; Galligan, J.J.; Hirschey, M.D.; Verdin, E.; Petersen, D.R. Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 2012, 11, 1633–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, B.D.; Tuma, D.J.; Tuma, P.L. Chronic ethanol consumption induces global hepatic protein hyperacetylation. Alcohol. Clin. Exp. Res. 2010, 34, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S.; Leo, M.A.; Wang, X.; Decarli, L.M. Alcohol alters hepatic FoxO1, p53, and mitochondrial SIRT5 deacetylation function. Biochem. Biophys. Res. Commun. 2008, 373, 246–252. [Google Scholar] [CrossRef]
- Picklo, M.J., Jr. Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem. Biophys. Res. Commun. 2008, 376, 615–619. [Google Scholar] [CrossRef]
- Marchner, H.; Tottmar, O. A comparative study on the effects of disulfiram, cyanamide and 1-aminocyclopropanol on the acetaldehyde metabolism in rats. Acta. Pharm. Toxicol. 1978, 43, 219–232. [Google Scholar] [CrossRef]
- Doody, E.E.; Groebner, J.L.; Walker, J.R.; Frizol, B.M.; Tuma, D.J.; Fernandez, D.J.; Tuma, P.L. Ethanol metabolism by alcohol dehydrogenase or cytochrome P450 2E1 differentially impairs hepatic protein trafficking and growth hormone signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G558–G569. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S.; DeCarli, L.M. The feeding of alcohol in liquid diets: Two decades of applications and 1982 update. Alcohol. Clin. Exp. Res. 1982, 6, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Ghosh Dastidar, S.; Warner, J.B.; Warner, D.R.; McClain, C.J.; Kirpich, I.A. Rodent Models of Alcoholic Liver Disease: Role of Binge Ethanol Administration. Biomolecules 2018, 8, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, B.D.; Tuma, P.L. Alcohol-induced protein hyperacetylation: Mechanisms and consequences. World J. Gastroenterol. 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, K.S.; Green, M.F.; Petersen, D.R.; Hirschey, M.D. Ethanol metabolism modifies hepatic protein acylation in mice. PLoS ONE 2013, 8, e75868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assiri, M.A.; Roy, S.R.; Harris, P.S.; Ali, H.; Liang, Y.; Shearn, C.T.; Orlicky, D.J.; Roede, J.R.; Hirschey, M.D.; Backos, D.S.; et al. Chronic Ethanol Metabolism Inhibits Hepatic Mitochondrial Superoxide Dismutase via Lysine Acetylation. Alcohol. Clin. Exp. Res. 2017, 41, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Gu, W. SIRT1: Regulator of p53 Deacetylation. Genes Cancer 2013, 4, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Duan, S.; Guan, T.; Yuan, X.; Lin, J.; Hou, S.; Lai, X.; Huang, S.; Du, X.; Chen, S. Vitexin protects against ethanol-induced liver injury through Sirt1/p53 signaling pathway. Eur. J. Pharm. 2020, 873, 173007. [Google Scholar] [CrossRef]
- Hu, M.; Wang, F.; Li, X.; Rogers, C.Q.; Liang, X.; Finck, B.N.; Mitra, M.S.; Zhang, R.; Mitchell, D.A.; You, M. Regulation of hepatic lipin-1 by ethanol: Role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology 2012, 55, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Ren, R.; Wang, Z.; Wu, M.; Wang, H. Emerging Roles of SIRT1 in Alcoholic Liver Disease. Int. J. Biol. Sci. 2020, 16, 3174–3183. [Google Scholar] [CrossRef]
- You, M.; Jogasuria, A.; Taylor, C.; Wu, J. Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 88–100. [Google Scholar] [CrossRef]
- Pandey, S.C.; Bohnsack, J.P. Alcohol Makes Its Epigenetic Marks. Cell Metab. 2020, 31, 213–214. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Shukla, S.D. Acute in vivo effect of ethanol (binge drinking) on histone H3 modifications in rat tissues. Alcohology 2006, 41, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.A.; Shepard, B.D.; Kannarkat, G.T.; Rutledge, T.M.; Tuma, D.J.; Tuma, P.L. Microtubule acetylation and stability may explain alcohol-induced alterations in hepatic protein trafficking. Hepatology 2008, 47, 1745–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.D.; Aroor, A.R.; Restrepo, R.; Kharbanda, K.K.; Ibdah, J.A. In Vivo Acute on Chronic Ethanol Effects in Liver: A Mouse Model Exhibiting Exacerbated Injury, Altered Metabolic and Epigenetic Responses. Biomolecules 2015, 5, 3280–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajmo, J.M.; Liang, X.; Rogers, C.Q.; Pennock, B.; You, M. Resveratrol alleviates alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G833–G842. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wan, T.; Ye, M.; Qiu, Y.; Pei, L.; Jiang, R.; Pang, N.; Huang, Y.; Liang, B.; Ling, W.; et al. Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1alpha/mitochondrial biosynthesis pathway. Redox Biol. 2018, 17, 89–98. [Google Scholar] [CrossRef]
- Assiri, M.A.; Ali, H.R.; Marentette, J.O.; Yun, Y.; Liu, J.; Hirschey, M.D.; Saba, L.M.; Harris, P.S.; Fritz, K.S. Investigating RNA expression profiles altered by nicotinamide mononucleotide therapy in a chronic model of alcoholic liver disease. Hum. Genom. 2019, 13, 65. [Google Scholar] [CrossRef]
- Pi, A.; Jiang, K.; Ding, Q.; Lai, S.; Yang, W.; Zhu, J.; Guo, R.; Fan, Y.; Chi, L.; Li, S. Alcohol Abstinence Rescues Hepatic Steatosis and Liver Injury via Improving Metabolic Reprogramming in Chronic Alcohol-Fed Mice. Front. Pharm. 2021, 12, 752148. [Google Scholar] [CrossRef]
- Donde, H.; Ghare, S.; Joshi-Barve, S.; Zhang, J.; Vadhanam, M.V.; Gobejishvili, L.; Lorkiewicz, P.; Srivastava, S.; McClain, C.J.; Barve, S. Tributyrin Inhibits Ethanol-Induced Epigenetic Repression of CPT-1A and Attenuates Hepatic Steatosis and Injury. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 569–585. [Google Scholar] [CrossRef] [Green Version]
- Barcena-Varela, M.; Lujambio, A. The Endless Sources of Hepatocellular Carcinoma Heterogeneity. Cancers 2021, 13, 2621. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Li, J.; Zheng, L.; Feng, M.; Wang, X.; Han, K.; Pi, H.; Li, M.; Huang, X.; et al. SIRT1 facilitates hepatocellular carcinoma metastasis by promoting PGC-1alpha-mediated mitochondrial biogenesis. Oncotarget 2016, 7, 29255–29274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Yazdani, H.O.; Liu, Y.; Loughran, P.; van der Windt, D.J.; Huang, H.; Simmons, R.L.; Shiva, S.; Tai, S.; Tsung, A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017, 66, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Nga, H.T.; Tian, J.; Yi, H.S. Mitochondrial Metabolic Signatures in Hepatocellular Carcinoma. Cells 2021, 10, 1901. [Google Scholar] [CrossRef]
- Choudhury, F.K. Mitochondrial Redox Metabolism: The Epicenter of Metabolism during Cancer Progression. Antioxidants 2021, 10, 1838. [Google Scholar] [CrossRef] [PubMed]
- De Matteis, S.; Granato, A.M.; Napolitano, R.; Molinari, C.; Valgiusti, M.; Santini, D.; Foschi, F.G.; Ercolani, G.; Vespasiani Gentilucci, U.; Faloppi, L.; et al. Interplay Between SIRT-3, Metabolism and Its Tumor Suppressor Role in Hepatocellular Carcinoma. Dig. Dis. Sci. 2017, 62, 1872–1880. [Google Scholar] [CrossRef]
- Hu, X.; Wang, D.; Sun, L.; Gao, Y.; Zhou, D.; Tong, X.; Li, J.; Lin, H.; Qing, Y.; Du, S.; et al. Disturbed mitochondrial acetylation in accordance with the availability of acetyl groups in hepatocellular carcinoma. Mitochondrion 2021, 60, 150–159. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Liu, L.; Cai, M.; Pan, Y.; Fu, J.; Cao, Y.; Yun, J. Low SIRT3 expression correlates with poor differentiation and unfavorable prognosis in primary hepatocellular carcinoma. PLoS ONE 2012, 7, e51703. [Google Scholar] [CrossRef]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012, 287, 42436–42443. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Wang, N.; Zhai, H.; Wang, R.; Wu, J.; Pu, W. SIRT3 functions as a tumor suppressor in hepatocellular carcinoma. Tumour Biol. 2017, 39, 1010428317691178. [Google Scholar] [CrossRef] [Green Version]
- Song, C.L.; Tang, H.; Ran, L.K.; Ko, B.C.; Zhang, Z.Z.; Chen, X.; Ren, J.H.; Tao, N.N.; Li, W.Y.; Huang, A.L.; et al. Sirtuin 3 inhibits hepatocellular carcinoma growth through the glycogen synthase kinase-3beta/BCL2-associated X protein-dependent apoptotic pathway. Oncogene 2016, 35, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, C.; Tian, Y.; Yao, Y.; Mao, J.; Wang, H.; Li, Z.; Xu, Y.; Ye, M.; Wang, L. SIRT5 Promotes Hepatocellular Carcinoma Progression by Regulating Mitochondrial Apoptosis. J. Cancer 2019, 10, 3871–3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Zhao, J.; Deng, W.; Chen, Y.; Shang, J.; Song, K.; Zhang, L.; Wang, C.; Lu, S.; Yang, X.; et al. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 2018, 14, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Laemmle, A.; Lechleiter, A.; Roh, V.; Schwarz, C.; Portmann, S.; Furer, C.; Keogh, A.; Tschan, M.P.; Candinas, D.; Vorburger, S.A.; et al. Inhibition of SIRT1 impairs the accumulation and transcriptional activity of HIF-1alpha protein under hypoxic conditions. PLoS ONE 2012, 7, e33433. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rodriguez, J.L.; Barbier-Torres, L.; Fernandez-Alvarez, S.; Gutierrez-de Juan, V.; Monte, M.J.; Halilbasic, E.; Herranz, D.; Alvarez, L.; Aspichueta, P.; Marin, J.J.; et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology 2014, 59, 1972–1983. [Google Scholar] [CrossRef] [Green Version]
- Karbasforooshan, H.; Hayes, A.W.; Mohammadzadeh, N.; Zirak, M.R.; Karimi, G. The possible role of Sirtuins and microRNAs in hepatocellular carcinoma therapy. Cell Cycle 2020, 19, 3209–3221. [Google Scholar] [CrossRef]
- Kim, J.K.; Noh, J.H.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Shen, Q.; Park, W.S.; Lee, J.Y.; et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology 2013, 57, 1055–1067. [Google Scholar] [CrossRef]
- Zhao, J.; Wozniak, A.; Adams, A.; Cox, J.; Vittal, A.; Voss, J.; Bridges, B.; Weinman, S.A.; Li, Z. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J. Exp. Clin. Cancer Res. 2019, 38, 252. [Google Scholar] [CrossRef] [Green Version]
- Jo, H.; Park, Y.; Kim, T.; Kim, J.; Lee, J.S.; Kim, S.Y.; Chung, J.I.; Ko, H.Y.; Pyun, J.C.; Kim, K.S.; et al. Modulation of SIRT3 expression through CDK4/6 enhances the anti-cancer effect of sorafenib in hepatocellular carcinoma cells. BMC Cancer 2020, 20, 332. [Google Scholar] [CrossRef] [Green Version]
- Tao, N.N.; Zhou, H.Z.; Tang, H.; Cai, X.F.; Zhang, W.L.; Ren, J.H.; Zhou, L.; Chen, X.; Chen, K.; Li, W.Y.; et al. Sirtuin 3 enhanced drug sensitivity of human hepatoma cells through glutathione S-transferase pi 1/JNK signaling pathway. Oncotarget 2016, 7, 50117–50130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Matteis, S.; Scarpi, E.; Granato, A.M.; Vespasiani-Gentilucci, U.; La Barba, G.; Foschi, F.G.; Bandini, E.; Ghetti, M.; Marisi, G.; Cravero, P.; et al. Role of SIRT-3, p-mTOR and HIF-1alpha in Hepatocellular Carcinoma Patients Affected by Metabolic Dysfunctions and in Chronic Treatment with Metformin. Int. J. Mol. Sci. 2019, 20, 1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Liu, Y.; Mi, X.; Chen, Q.; Jiang, P.; Hou, J.; Lin, Y.; Li, S.; Ji, B.; Fang, Y. Sirt3 promotes hepatocellular carcinoma cells sensitivity to regorafenib through the acceleration of mitochondrial dysfunction. Arch. Biochem. Biophys. 2020, 689, 108415. [Google Scholar] [CrossRef]
- Xia, Y.Q.; Hua, R.J.; Juan, C.; Zhong, Z.H.; Tao, C.S.; Fang, R.; Lin, H.; Rui, G.; Yong, C. SIRT6 Depletion Sensitizes Human Hepatoma Cells to Chemotherapeutics by Downregulating MDR1 Expression. Front. Pharm. 2018, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhang, B.; Wong, N.; Lo, A.W.; To, K.F.; Chan, A.W.; Ng, M.H.; Ho, C.Y.; Cheng, S.H.; Lai, P.B.; et al. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res. 2011, 71, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chan, A.W.; To, K.F.; Chen, W.; Zhang, Z.; Ren, J.; Song, C.; Cheung, Y.S.; Lai, P.B.; Cheng, S.H.; et al. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3beta/beta-catenin signaling. Hepatology 2013, 57, 2287–2298. [Google Scholar] [CrossRef]
- Nancy Han, M.; Rong Wei, P. Age-Adjusted Death Rates for Alzheimer Disease Among Adults Aged ≤ 65 Years, by Sex—National Vital Statistics System, United States, 1999–2019. MMWR Morb. Mortal Wkly. 2021, 70, 602. [Google Scholar] [CrossRef]
- Yen, T.C.; Chen, Y.S.; King, K.L.; Yeh, S.H.; Wei, Y.H. Liver Mitochondrial Respiratory Functions Decline with Age. Biochem. Biophys. Res. Commun. 1989, 165, 994–1003. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Argmann, C.; Houten, S.M.; Canto, C.; Jeninga, E.H.; Andreux, P.A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. The metabolic footprint of aging in mice. Sci. Rep. 2011, 1, 134. [Google Scholar] [CrossRef]
- Bradshaw, P.C. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Mitochondrial acetylation and diseases of aging. J. Aging Res. 2011, 2011, 234875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldensperger, T.; Eggen, M.; Kappen, J.; Winterhalter, P.R.; Pfirrmann, T.; Glomb, M.A. Comprehensive analysis of posttranslational protein modifications in aging of subcellular compartments. Sci. Rep. 2020, 10, 7596. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Green, M.F.; Huynh, F.K.; Wagner, G.R.; Hirschey, M.D. SnapShot: Mammalian Sirtuins. Cell 2014, 159, 956–956.e1. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef]
- Nahalkova, J. Focus on Molecular Functions of Anti-Aging Deacetylase SIRT3. Biochemistry 2022, 87, 21–34. [Google Scholar] [CrossRef]
- Lee, J.Y.; Hong, S.H. Hematopoietic Stem Cells and Their Roles in Tissue Regeneration. Int. J. Stem Cells 2020, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Roichman, A.; Elhanati, S.; Aon, M.A.; Abramovich, I.; Di Francesco, A.; Shahar, Y.; Avivi, M.Y.; Shurgi, M.; Rubinstein, A.; Wiesner, Y.; et al. Restoration of energy homeostasis by SIRT6 extends healthy lifespan. Nat. Commun. 2021, 12, 3208. [Google Scholar] [CrossRef]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.H.; Lu, M.; Lee, B.Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef] [PubMed]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherji, A.; Bailey, S.M.; Staels, B.; Baumert, T.F. The circadian clock and liver function in health and disease. J. Hepatol. 2019, 71, 200–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Solanas, G.; Peixoto, F.O.; Bee, L.; Symeonidi, A.; Schmidt, M.S.; Brenner, C.; Masri, S.; Benitah, S.A.; Sassone-Corsi, P. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 2017, 170, 664–677.e11. [Google Scholar] [CrossRef]
- Levine, D.C.; Hong, H.; Weidemann, B.J.; Ramsey, K.M.; Affinati, A.H.; Schmidt, M.S.; Cedernaes, J.; Omura, C.; Braun, R.; Lee, C.; et al. NAD(+) Controls Circadian Reprogramming through PER2 Nuclear Translocation to Counter Aging. Mol. Cell 2020, 78, 835–849.e7. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E.F. Therapeutic potential of boosting NAD+ in aging and age-related diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Intervention | System | MOA | Outcomes | Ref. |
| MetS | Resveratrol | 41 overweight men and women: double-blind clinical trial. 150 mg/d of resveratrol (n = 20) or placebo (n = 21) for 6 months (NCT02565979). 74 men with MetS: 3 groups. 1000 mg/d resveratrol (n = 21), 150 mg/d resveratrol (n = 21) and placebo (n = 24) for 16 weeks (NCT01412645). | SIRT1 agonist | No difference was observed in insulin sensitivity, body composition, blood pressure, energy metabolism, but there was a decrease in glycated hemoglobin. No improvement in inflammatory status, glucose homeostasis, plod pressure or hepatic lipid content. | [131,132] |
| Time-restricted feeding | 30 participants with metabolic syndrome. Restricted eating to 10 hours a day. 344 participants with type 2 diabetes: 4 groups. No change to meal timing or light exposure; early time-restricted feeding; light therapy; early time-restricted feeding and light therapy (NCT04155619). | Improve circadian rhythms, increase expression of AMPK, FOXO, sirtuins, and ketogenesis. | Publication pending. | [135,136] | |
| Metformin | 50 patients: 3 groups. All treated with hypocaloric diet. Obese normoglycemic (n = 18); pre- diabetic treated with metformin (n = 16); pre-DM obese patients treated with metformin (n = 16). | Increased SIRT6 expression and decreased NF-κB, PPAR, SREBP1 expression in obese pre-diabetic patients treated with metformin. | [118] | ||
| NR | 40 male participants: 2 groups. 1 g nicotinamide riboside twice daily; placebo for 12 weeks (NCT02303483). | Increase NAD+ | Failed to improve insulin sensitivity, whole-body glucose, body composition in obese and insulin- resistant men. | [137] | |
| NAFLD | Resveratrol | Patients with NAFLD (n = 50): double blind clinical trial. 500 mg/d resveratrol (n = 25) or placebo (n = 25) for 12 weeks (NCT02030977). | SIRT1 agonist | Change in hepatic steatosis and inflammation | [102] |
| Nicotinamide | 61 participants: 2 groups. Both groups receiving anti-diabetic therapy (control, n = 30) with one supplemented with 1g/d nicotinamide (n = 31) for 12 weeks (NCT03850886). | Increase NAD+ | Improved steatosis score, metabolic abnormalities and quality of life but failed to diminish liver fibrosis or steatosis. | [138] | |
| NMN | 25 female participants: double-blind trail, 2 groups. 250 mg/d of NMN (n = 13) or placebo (n = 12) for 10 weeks (NCT03151239). | Increase NAD+ | Improves skeletal muscle insulin signaling, insulin sensitivity and muscle remodeling in post-menopausal prediabetic women. | [139] | |
| Time-restricted Feeding | 88 participants: 2 groups. Time restricted feeding (8 hour feeding window); Continuous energy restriction (NCT03786523). | Publication pending. | |||
| Cancer | Time-restricted Eating | 40 participants: single group assignment. Time-restricted eating (10 hour feeding window) for 14 days (NCT04243512). | Reduced fatigue. | [135,140] | |
| Aging | NMN | 66 participants: 2 groups. 300 mg/d NMN or placebo for 60 days (NCT04228640). 90 participants: 6 groups. 300 mg/d NMN (n = 20) or placebo (n = 7); 600 mg/d NMN (n = 20) or placebo (n = 7); 900 mg/d NMN (n = 10) or placebo (n = 6) for 60 days (NCT04823260). | Increase NAD+ | Significantly elongated telomere length in lymphocytes, monocytes, and dendritic cells. Publication pending. | [141] |
| Caloric Restriction | 71 participants: 3 groups. Calorie restricting (n = 30); normal-eating controls (n = 16); obese comparison (n = 25) (NCT01256840). | Did not show delayed aging in either telomere length or T cell senescence markers. | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGinnis, C.D.; Jennings, E.Q.; Harris, P.S.; Galligan, J.J.; Fritz, K.S. Biochemical Mechanisms of Sirtuin-Directed Protein Acylation in Hepatic Pathologies of Mitochondrial Dysfunction. Cells 2022, 11, 2045. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11132045
McGinnis CD, Jennings EQ, Harris PS, Galligan JJ, Fritz KS. Biochemical Mechanisms of Sirtuin-Directed Protein Acylation in Hepatic Pathologies of Mitochondrial Dysfunction. Cells. 2022; 11(13):2045. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11132045
Chicago/Turabian StyleMcGinnis, Courtney D., Erin Q. Jennings, Peter S. Harris, James J. Galligan, and Kristofer S. Fritz. 2022. "Biochemical Mechanisms of Sirtuin-Directed Protein Acylation in Hepatic Pathologies of Mitochondrial Dysfunction" Cells 11, no. 13: 2045. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11132045