
Inhibition of Mcl-1 Synergistically Enhances the Antileukemic Activity of Gilteritinib and MRX-2843 in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Culture
2.3. Clinical Samples
2.4. Annexin V/Propidium Iodide (PI) Staining and Flow Cytometry Analyses
2.5. Western Blots
2.6. Mitochondrial Fractionation/Cytochrome c Release
2.7. shRNA Knockdown of Bak and Bax
2.8. CRISPR Knockdown
2.9. Colony-Forming Assay
2.10. Real-Time RT-PCR
2.11. Statistical Analysis
3. Results
3.1. AZD5991 Synergistically Enhances Apoptosis Induced by FLT3 Inhibition in FLT3-Mutated AML Cells
3.2. AZD5991 Alone and in Combination with Gilteritinib and MRX-2843 Significantly Reduces Colony-Forming Capacity of FLT3-Mutated AML Progenitor Cells
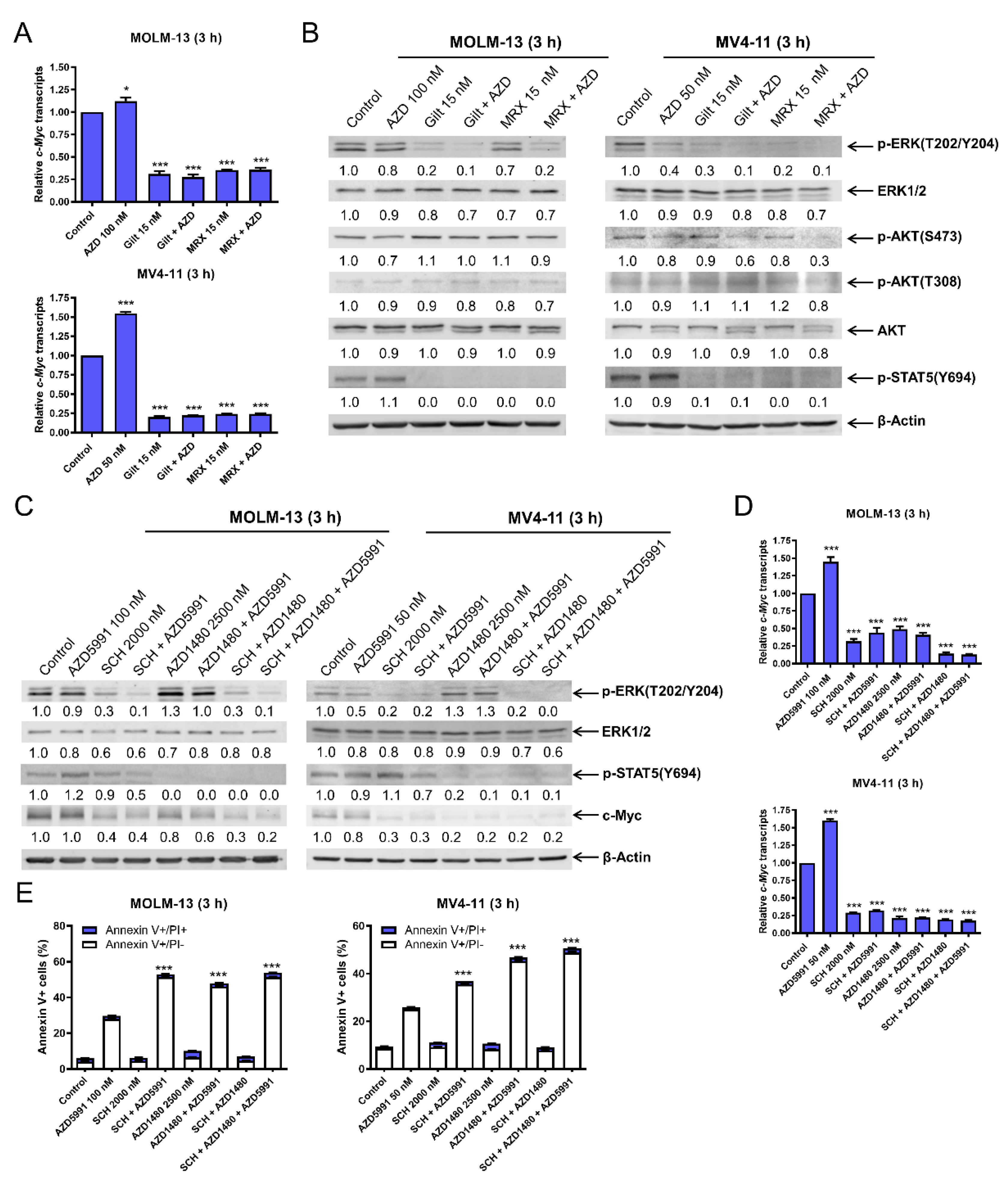
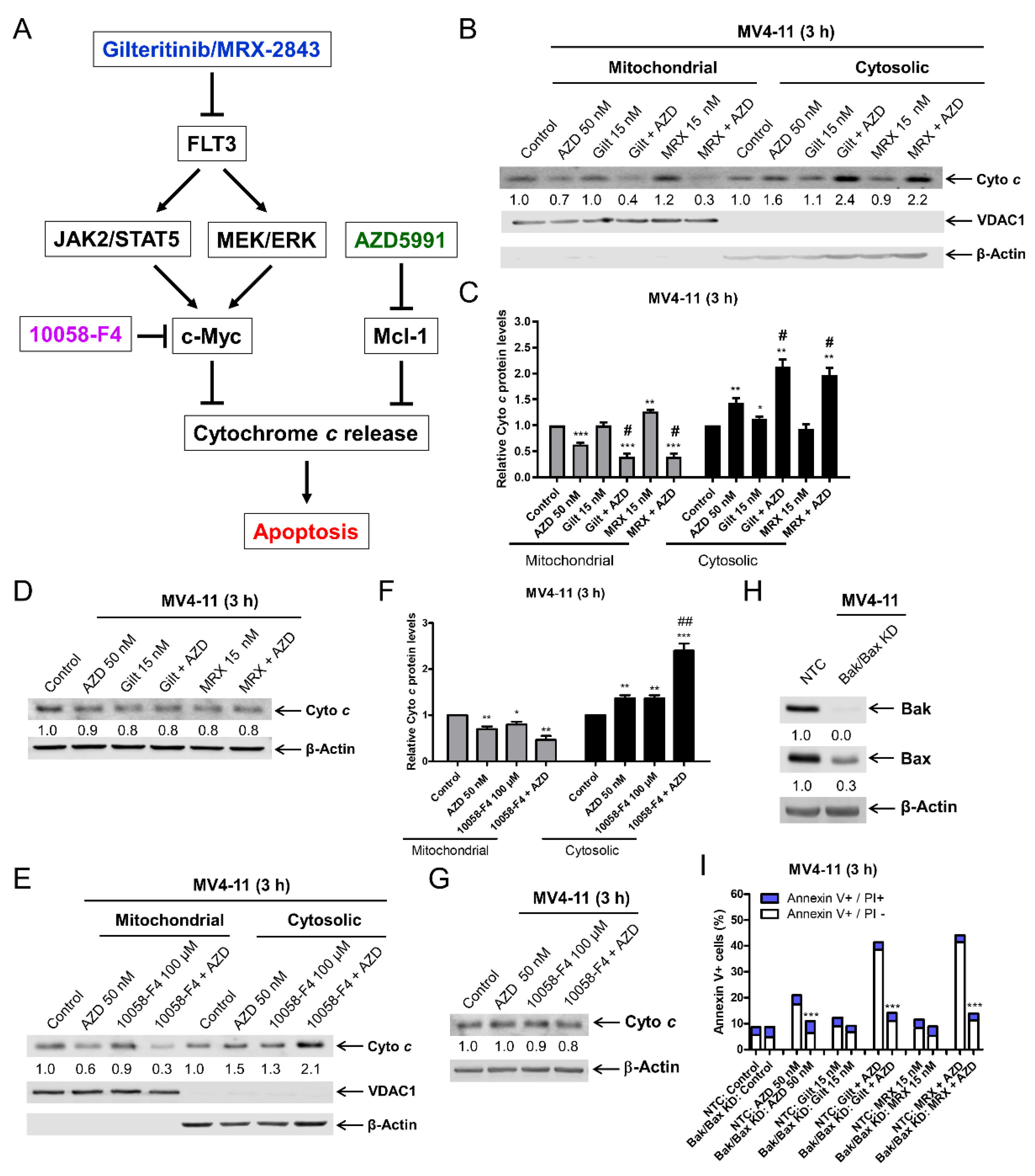
3.3. c-Myc Plays an Important Role in the Synergistic Antileukemic Activity of AZD5991 and Gilteritinib or MRX-2843 in FLT3-Mutated AML Cells
3.4. FLT3 Inhibition Decreases c-Myc Protein Level via Transcriptional Regulation through Suppression of the MEK/ERK and JAK2/STAT5 Pathways
3.5. FLT3 Inhibition Enhances AZD5991-Induced Cytochrome c Release
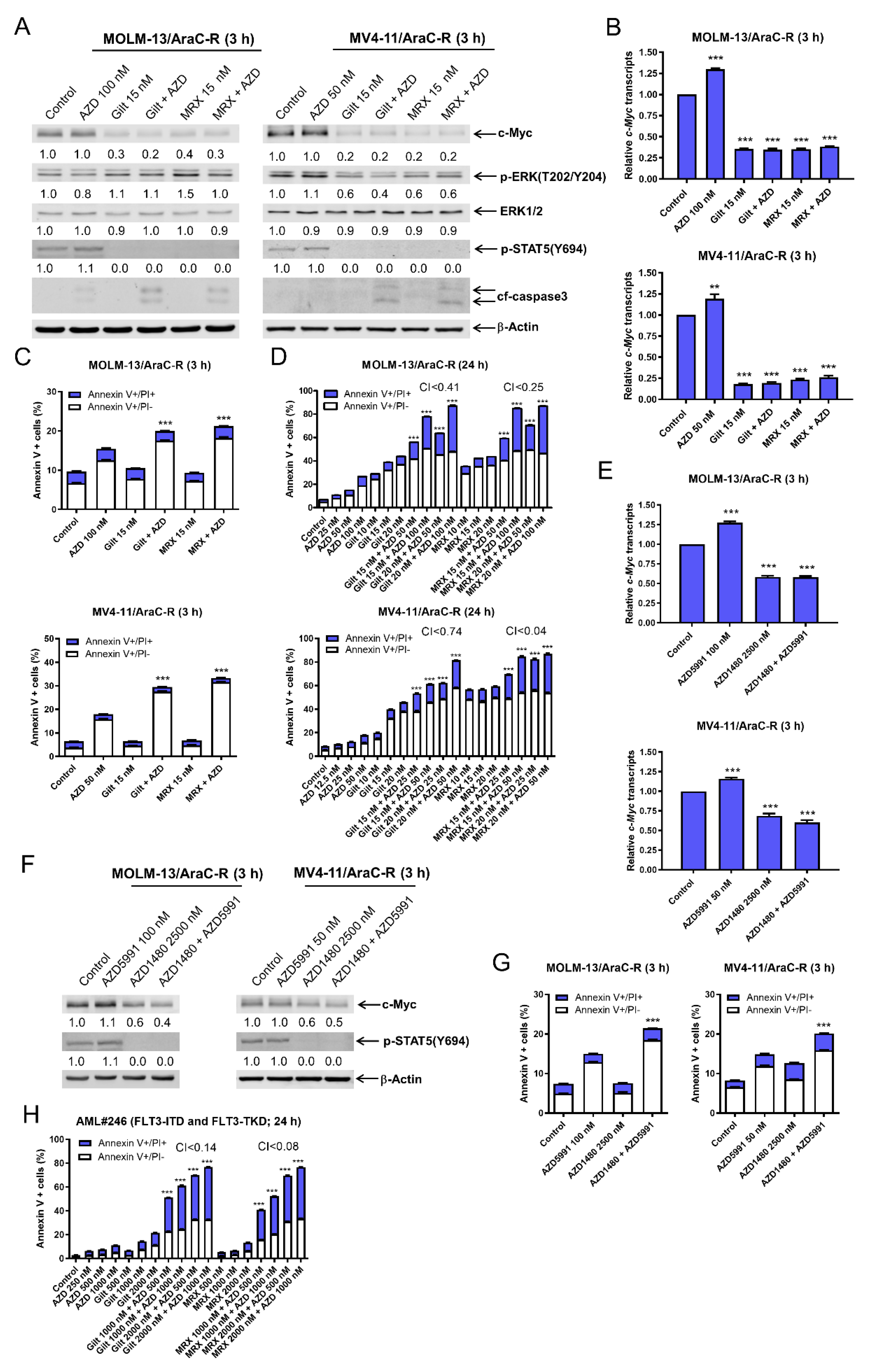
3.6. AZD5991 Synergistically Enhances Apoptosis Induced by Gilteritinib and MRX-2843 in AraC Resistant AML Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed]
- Deschler, B.; Lubbert, M. Acute myeloid leukemia: Epidemiology and etiology. Cancer 2006, 107, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research, N.; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Yamaguchi, H.; Kuboyama, M.; Najima, Y.; Usuki, K.; Ueki, T.; Oh, I.; Mori, S.; Kawata, E.; Uoshima, N.; et al. Significance of FLT3-tyrosine kinase domain mutation as a prognostic factor for acute myeloid leukemia. Int. J. Hematol. 2019, 110, 566–574. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters--an analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Knight, T.E.; Edwards, H.; Meshinchi, S.; Taub, J.W.; Ge, Y. “FLipping”; the Story: FLT3-Mutated Acute Myeloid Leukemia and the Evolving Role of FLT3 Inhibitors. Cancers 2022, 14, 3398. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; DeRyckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef]
- Lee-Sherick, A.B.; Eisenman, K.M.; Sather, S.; McGranahan, A.; Armistead, P.M.; McGary, C.S.; Hunsucker, S.A.; Schlegel, J.; Martinson, H.; Cannon, C.; et al. Aberrant Mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene 2013, 32, 5359–5368. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Okada, K.; Nogami, A.; Ishida, S.; Akiyama, H.; Chen, C.; Umezawa, Y.; Miura, O. FLT3-ITD induces expression of Pim kinases through STAT5 to confer resistance to the PI3K/Akt pathway inhibitors on leukemic cells by enhancing the mTORC1/Mcl-1 pathway. Oncotarget 2018, 9, 8870–8886. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Ma, J.; Knight, T.; Su, Y.; Edwards, H.; Polin, L.; Li, J.; Kushner, J.; Dzinic, S.H.; White, K.; et al. The combination of CUDC-907 and gilteritinib shows promising in vitro and in vivo antileukemic activity against FLT3-ITD AML. Blood Cancer J. 2021, 11, 111. [Google Scholar] [CrossRef]
- Liu, S.; Qiao, X.; Wu, S.; Gai, Y.; Su, Y.; Edwards, H.; Wang, Y.; Lin, H.; Taub, J.W.; Wang, G.; et al. c-Myc Plays a Critical Role in the Antileukemic Activity of the Mcl-1-selective Inhibitor AZD5991 in Acute Myeloid Leukemia. Apoptosis, 2022; online ahead of print. [Google Scholar]
- Kelly, G.L.; Grabow, S.; Glaser, S.P.; Fitzsimmons, L.; Aubrey, B.J.; Okamoto, T.; Valente, L.J.; Robati, M.; Tai, L.; Fairlie, W.D.; et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev. 2014, 28, 58–70. [Google Scholar] [CrossRef]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.S.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef]
- Xiang, Z.; Luo, H.; Payton, J.E.; Cain, J.; Ley, T.J.; Opferman, J.T.; Tomasson, M.H. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J. Clin. Invest. 2010, 120, 2109–2118. [Google Scholar] [CrossRef]
- Zhao, J.; Niu, X.; Li, X.; Edwards, H.; Wang, G.; Wang, Y.; Taub, J.W.; Lin, H.; Ge, Y. Inhibition of CHK1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Oncotarget 2016, 7, 34785–34799. [Google Scholar] [CrossRef]
- Uphoff, C.C.; Drexler, H.G. Detection of mycoplasma contaminations. Methods Mol. Biol. 2005, 290, 13–23. [Google Scholar]
- Niu, X.; Wang, G.; Wang, Y.; Caldwell, J.T.; Edwards, H.; Xie, C.; Taub, J.W.; Li, C.; Lin, H.; Ge, Y. Acute myeloid leukemia cells harboring MLL fusion genes or with the acute promyelocytic leukemia phenotype are sensitive to the Bcl-2-selective inhibitor ABT-199. Leukemia 2014, 28, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, X.; Su, Y.; Zhao, J.; Luedtke, D.A.; Epshteyn, V.; Edwards, H.; Wang, G.; Wang, Z.; Chu, R.; et al. Mechanisms responsible for the synergistic antileukemic interactions between ATR inhibition and cytarabine in acute myeloid leukemia cells. Sci. Rep. 2017, 7, 41950. [Google Scholar] [CrossRef]
- Taub, J.W.; Matherly, L.H.; Stout, M.L.; Buck, S.A.; Gurney, J.G.; Ravindranath, Y. Enhanced metabolism of 1-beta-D-arabinofuranosylcytosine in Down Syndrome cells: A contributing factor to the superior event free survival of Down Syndrome children with acute myeloid leukemia. Blood 1996, 87, 3395–3403. [Google Scholar] [CrossRef]
- Xie, C.; Edwards, H.; Xu, X.; Zhou, H.; Buck, S.A.; Stout, M.L.; Yu, Q.; Rubnitz, J.E.; Matherly, L.H.; Taub, J.W.; et al. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5499–5510. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Stout, M.L.; Tatman, D.A.; Jensen, T.L.; Buck, S.; Thomas, R.L.; Ravindranath, Y.; Matherly, L.H.; Taub, J.W. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J. Natl. Cancer Inst. 2005, 97, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Dombkowski, A.A.; LaFiura, K.M.; Tatman, D.; Yedidi, R.S.; Stout, M.L.; Buck, S.A.; Massey, G.; Becton, D.L.; Weinstein, H.J.; et al. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood 2006, 107, 1570–1581. [Google Scholar] [CrossRef]
- Liu, F.; Kalpage, H.A.; Wang, D.; Edwards, H.; Hüttemann, M.; Ma, J.; Su, Y.; Carter, J.; Li, X.; Polin, L.; et al. Cotargeting of Mitochondrial Complex I and Bcl-2 Shows Antileukemic Activity against Acute Myeloid Leukemia Cells Reliant on Oxidative Phosphorylation. Cancers 2020, 12, 2400. [Google Scholar] [CrossRef]
- Xie, C.; Drenberg, C.; Edwards, H.; Caldwell, J.T.; Chen, W.; Inaba, H.; Xu, X.; Buck, S.A.; Taub, J.W.; Baker, S.D.; et al. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PloS ONE 2013, 8, e79106. [Google Scholar] [CrossRef]
- Luedtke, D.A.; Su, Y.; Ma, J.; Li, X.; Buck, S.A.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; et al. Inhibition of CDK9 by voruciclib synergistically enhances cell death induced by the Bcl-2 selective inhibitor venetoclax in preclinical models of acute myeloid leukemia. Signal Transduct. Target. Ther. 2020, 5, 17. [Google Scholar] [CrossRef]
- Li, X.; Su, Y.; Madlambayan, G.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Ma, J.; Knight, T.; et al. Antileukemic activity and mechanism of action of the novel PI3K and histone deacetylase dual inhibitor CUDC-907 in acute myeloid leukemia. Haematologica 2019, 104, 2225–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Li, X.; Ma, J.; Zhao, J.; Liu, S.; Wang, G.; Edwards, H.; Taub, J.W.; Lin, H.; Ge, Y. Targeting PI3K, mTOR, ERK, and Bcl-2 signaling network shows superior antileukemic activity against AML ex vivo. Biochem. Pharmacol. 2018, 148, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Luedtke, D.A.; Su, Y.; Liu, S.; Edwards, H.; Wang, Y.; Lin, H.; Taub, J.W.; Ge, Y. Inhibition of XPO1 enhances cell death induced by ABT-199 in acute myeloid leukaemia via Mcl-1. J. Cell Mol. Med. 2018, 22, 6099–6111. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Juin, P.; Hueber, A.O.; Littlewood, T.; Evan, G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999, 13, 1367–1381. [Google Scholar] [CrossRef]
- Singh Mali, R.; Zhang, Q.; DeFilippis, R.A.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2021, 106, 1034–1046. [Google Scholar] [CrossRef]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Piccotti, L.; Buratta, M.; Giannini, S.; Gresele, P.; Roberti, R.; Corazzi, L. Binding and release of cytochrome c in brain mitochondria is influenced by membrane potential and hydrophobic interactions with cardiolipin. J. Membr. Biol. 2004, 198, 43–53. [Google Scholar] [CrossRef]
- Carter, B.Z.; Mak, P.Y.; Tao, W.; Warmoes, M.; Lorenzi, P.L.; Mak, D.; Ruvolo, V.; Tan, L.; Cidado, J.; Drew, L.; et al. Targeting MCL-1 dysregulates cell metabolism and leukemia-stroma interactions and resensitizes acute myeloid leukemia to BCL-2 inhibition. Haematologica 2022, 107, 58–76. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Zavorka Thomas, M.E.; Lu, X.; Talebi, Z.; Jeon, J.Y.; Buelow, D.R.; Gibson, A.A.; Uddin, M.E.; Brinton, L.T.; Nguyen, J.; Collins, M.; et al. Gilteritinib Inhibits Glutamine Uptake and Utilization in FLT3-ITD-Positive AML. Mol. Cancer Ther. 2021, 20, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.; Amanullah, A.; Shafarenko, M.; Liebermann, D.A. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene 2002, 21, 3414–3421. [Google Scholar] [CrossRef]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle, A.; León, J. MYC oncogene in myeloid neoplasias. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2013, 15, 87–94. [Google Scholar] [CrossRef]
- Mughal, M.K.; Akhter, A.; Street, L.; Pournazari, P.; Shabani-Rad, M.T.; Mansoor, A. Acute myeloid leukaemia: Expression of MYC protein and its association with cytogenetic risk profile and overall survival. Hematol. Oncol. 2017, 35, 350–356. [Google Scholar] [CrossRef]
- Benetatos, L.; Benetatou, A.; Vartholomatos, G. Long non-coding RNAs and MYC association in hematological malignancies. Ann. Hematol. 2020, 99, 2231–2242. [Google Scholar] [CrossRef]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. CR 2018, 37, 173. [Google Scholar] [CrossRef]
- Pan, X.N.; Chen, J.J.; Wang, L.X.; Xiao, R.Z.; Liu, L.L.; Fang, Z.G.; Liu, Q.; Long, Z.J.; Lin, D.J. Inhibition of c-Myc overcomes cytotoxic drug resistance in acute myeloid leukemia cells by promoting differentiation. PloS ONE 2014, 9, e105381. [Google Scholar] [CrossRef]
- Mudgapalli, N.; Nallasamy, P.; Chava, H.; Chava, S.; Pathania, A.S.; Gunda, V.; Gorantla, S.; Pandey, M.K.; Gupta, S.C.; Challagundla, K.B. The role of exosomes and MYC in therapy resistance of acute myeloid leukemia: Challenges and opportunities. Mol. Asp. Med. 2019, 70, 21–32. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.; Edwards, H.; Wang, D.; Liu, S.; Qiao, X.; Carter, J.; Wang, Y.; Taub, J.W.; Wang, G.; Ge, Y. Inhibition of Mcl-1 Synergistically Enhances the Antileukemic Activity of Gilteritinib and MRX-2843 in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Cells 2022, 11, 2752. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172752
Wu S, Edwards H, Wang D, Liu S, Qiao X, Carter J, Wang Y, Taub JW, Wang G, Ge Y. Inhibition of Mcl-1 Synergistically Enhances the Antileukemic Activity of Gilteritinib and MRX-2843 in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Cells. 2022; 11(17):2752. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172752
Chicago/Turabian StyleWu, Shuangshuang, Holly Edwards, Deying Wang, Shuang Liu, Xinan Qiao, Jenna Carter, Yue Wang, Jeffrey W. Taub, Guan Wang, and Yubin Ge. 2022. "Inhibition of Mcl-1 Synergistically Enhances the Antileukemic Activity of Gilteritinib and MRX-2843 in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia" Cells 11, no. 17: 2752. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172752