
Na,K-ATPase Acts as a Beta-Amyloid Receptor Triggering Src Kinase Activation
 , , , ,
, , , ,  ,
,  , ,
, ,
Abstract
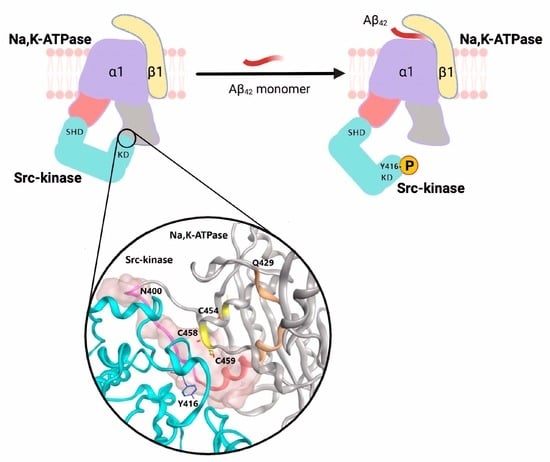
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Aβ42 Preparation
2.3. pNaKtide Preparation
2.4. Co-Localization Studies with Confocal Microscopy
2.4.1. Cell Staining for the β1-Subunit of Na,K-ATPase and Aβ42
2.4.2. Cell Staining for the α1-Subunit of Na,K-ATPase and Aβ42
2.4.3. Laser Scanning Confocal Microscopy
2.5. Co-Localization Studies with Proximity Ligation Assay
2.5.1. Primary Immunolabeling of Aβ42 and Na,K-ATPase
2.5.2. Primary Immunolabeling of Src Kinase and Na,K-ATPase
2.5.3. Fluorescent and Confocal Imaging for Proximity Ligation Assay
- -
- Hoechst fluorescence (DNA staining) with excitation at 405 nm and emission at 414–487 nm;
- -
- RNASelect Green fluorescence, excitation at 488 nm, emission at 495–590 nm;
- -
- Duolink Detection Reagent (Red) fluorescence, excitation at 594 nm, emission at 600–652 nm.
2.6. Assessment of the Level of Na,K-ATPase and Redox Status of the Cells
2.7. Analysis of Src Kinase Phosphorylation Levels in SH-SY5Y Cells
2.8. Phosphorylation of Src Kinase In Vitro
2.9. Microscale Thermophoresis
2.10. Measurement of Na,K-ATPase Transport Activity by Atomic Adsorption Spectrometry
2.11. Modeling the Interaction of Human Src Kinase with the Nucleotide Binding Domain of β1-Subunit of Human Na,K-ATPase
2.12. Statistical Analysis
3. Results
3.1. Aβ42 Co-Localizes with Na,K-ATPase and Initiates Src Signaling
3.2. Activation of Src Kinase by Aβ42 Is Mediated by Na,K-ATPase
3.3. Aβ42 Affects Cellular Redox-State and Does Not Activate Src Kinase under Hypoxia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Cell biology. A unifying role for prions in neurodegenerative diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. What is the Physiological Function of Amyloid-Beta Protein? J. Nutr. Health Aging 2019, 23, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Quintela-López, T.; Ortiz-Sanz, C.; Serrano-Regal, M.P.; Gaminde-Blasco, A.; Valero, J.; Baleriola, J.; Sбnchez-Gymez, M.V.; Matute, C.; Alberdi, E. Aβ oligomers promote oligodendrocyte differentiation and maturation via integrin β1 and Fyn kinase signaling. Cell Death Dis. 2019, 10, 445. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 14537–14545. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Privitera, L.; Fa’, M.; Staniszewski, A.; Hashimoto, G.; Aziz, F.; Sakurai, M.; Ribe, E.M.; Troy, C.M.; Mercken, M.; et al. Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Ann. Neurol. 2011, 69, 819–830. [Google Scholar] [CrossRef]
- Dhawan, G.; Combs, C.K. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 117. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.-L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.; Portugal, C.C.; Canedo, T.; Rodrigues, A.; Almeida, T.O.; Henriques, J.F.; Vaz, S.H.; Magalhães, J.; Silva, C.M.; Baptista, F.I.; et al. Microglia Dysfunction Caused by the Loss of Rhoa Disrupts Neuronal Physiology and Leads to Neurodegeneration. Cell Rep. 2020, 31, 107796. [Google Scholar] [CrossRef] [PubMed]
- Portugal, C.C.; Almeida, T.O.; Socodato, R.; Relvas, J.B. Src family kinases (SFKs): Critical regulators of microglial homeostatic functions and neurodegeneration in Parkinson’s and Alzheimer’s diseases. FEBS J. 2021; early view. [Google Scholar] [CrossRef]
- Dickey, C.A.; Gordon, M.N.; Wilcock, D.M.; Herber, D.L.; Freeman, M.J.; Morgan, D. Dysregulation of Na+/K+ ATPase by amyloid in APP + PS1 transgenic mice. BMC Neurosci. 2005, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Mitkevich, V.A.; Anashkina, A.A.; Adzhubei, A.A.; Burnysheva, K.M.; Lakunina, V.A.; Kamanina, Y.V.; Dergousova, E.A.; Lopina, O.D.; Ogunshola, O.O.; et al. Direct interaction of beta-amyloid with Na,K-ATPase as a putative regulator of the enzyme function. Sci. Rep. 2016, 6, 27738. [Google Scholar] [CrossRef]
- Kreutz, F.; Scherer, E.B.; Ferreira, A.G.K.; Petry, F.D.S.; Pereira, C.L.; Santana, F.; de Souza Wyse, A.T.; Salbego, C.G.; Trindade, V.M.T. Alterations on Na+,K+-ATPase and acetylcholinesterase activities induced by amyloid-β peptide in rat brain and GM1 ganglioside neuroprotective action. Neurochem. Res. 2013, 38, 2342–2350. [Google Scholar] [CrossRef]
- Ohnishi, T.; Yanazawa, M.; Sasahara, T.; Kitamura, Y.; Hiroaki, H.; Fukazawa, Y.; Kii, I.; Nishiyama, T.; Kakita, A.; Takeda, H.; et al. Na,K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc. Natl. Acad. Sci. USA. 2015, 112, E4465–E4474. [Google Scholar] [CrossRef]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11, 302. [Google Scholar] [CrossRef]
- Zhang, L.-N.; Sun, Y.-J.; Pan, S.; Li, J.-X.; Qu, Y.-E.; Li, Y.; Wang, Y.-L.; Gao, Z.-B. Na+-K+-ATPase, a potent neuroprotective modulator against Alzheimer disease. Fundam. Clin. Pharmacol. 2013, 27, 96–103. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-amyloid monomers are neuroprotective. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef]
- Bate, C.; Williams, A. Monomeric amyloid-β reduced amyloid-β oligomer-induced synapse damage in neuronal cultures. Neurobiol. Dis. 2018, 111, 48–58. [Google Scholar] [CrossRef]
- Yu, H.; Cui, X.; Zhang, J.; Xie, J.X.; Banerjee, M.; Pierre, S.V.; Xie, Z. Heterogeneity of signal transduction by Na-K-ATPase α-isoforms: Role of Src interaction. Am. J. Physiol. Cell Physiol. 2018, 314, C202–C210. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Askari, A. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.-Y.; Xie, Z.-J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Duan, Q.; Xie, Z. SH2 Ligand-Like Effects of Second Cytosolic Domain of Na/K-ATPase α1 Subunit on Src Kinase. PLoS ONE 2015, 10, e0142119. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Cui, X.; Li, Z.; Yu, H.; Cai, L.; Jia, X.; He, D.; Wang, C.; Gao, T.; Xie, Z. Na/K-ATPase Y260 Phosphorylation-mediated Src Regulation in Control of Aerobic Glycolysis and Tumor Growth. Sci. Rep. 2018, 8, 12322. [Google Scholar] [CrossRef]
- Nie, Y.; Bai, F.; Chaudhry, M.A.; Pratt, R.; Shapiro, J.I.; Liu, J. The Na/K-ATPase α1 and c-Src form signaling complex under native condition: A crosslinking approach. Sci. Rep. 2020, 10, 6006. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.; Scales, T.; Clark, B.R.; Gibb, G.; Reynolds, C.H.; Kellie, S.; Bird, I.N.; Varndell, I.M.; Sheppard, P.W.; Everall, I.; et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: Involvement of Src family protein kinases. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 10–20. [Google Scholar] [CrossRef]
- Klein, W.L. Abeta toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [Green Version]
- Gullberg, M.; Göransson, C.; Fredriksson, S. Duolink-“In-cell Co-IP” for visualization of protein interactions in situ. Nat. Methods 2011, 8, i–ii. [Google Scholar] [CrossRef]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef]
- Anashkina, A.A.; Kravatsky, Y.; Kuznetsov, E.; Makarov, A.A.; Adzhubei, A.A. Meta-server for automatic analysis, scoring and ranking of docking models. Bioinforma. Oxf. Engl. 2018, 34, 297–299. [Google Scholar] [CrossRef]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Liang, M.; Cai, T.; Tian, J.; Qu, W.; Xie, Z.-J. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. J. Biol. Chem. 2006, 281, 19709–19719. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Mitkevich, V.A.; Makarov, A.A. Molecular Mechanisms of the Redox Regulation of the Na,K-ATPase. Biophysics 2020, 65, 711–730. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Liu, J.; Yuan, Z.; Pierre, S.V.; Qu, W.; Zhao, X.; Xie, Z. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic. Biol. Med. 2006, 41, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol. Cell. Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Mitkevich, V.A.; Lakunina, V.A.; Anashkina, A.A.; Spirin, P.V.; Rubtsov, P.M.; Prassolov, V.S.; Bogdanov, N.B.; Hänggi, P.; Fuller, W.; et al. Cysteine residues 244 and 458–459 within the catalytic subunit of Na,K-ATPase control the enzyme’s hydrolytic and signaling function under hypoxic conditions. Redox Biol. 2017, 13, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Giedraitis, V.; Sundelöf, J.; Irizarry, M.C.; Gårevik, N.; Hyman, B.T.; Wahlund, L.-O.; Ingelsson, M.; Lannfelt, L. The normal equilibrium between CSF and plasma amyloid beta levels is disrupted in Alzheimer’s disease. Neurosci. Lett. 2007, 427, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.-M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef] [Green Version]
- Orlov, S.N.; Tverskoi, A.M.; Sidorenko, S.V.; Smolyaninova, L.V.; Lopina, O.D.; Dulin, N.O.; Klimanova, E.A. Na,K-ATPase as a target for endogenous cardiotonic steroids: What’s the evidence? Genes Dis. 2021, 8, 259–271. [Google Scholar] [CrossRef]
- Pavlovic, D. Endogenous cardiotonic steroids and cardiovascular disease, where to next? Cell Calcium 2020, 86, 102156. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Fedorova, O.V.; Dmitrieva, R.I.; Howald, W.N.; Hunter, A.P.; Kuznetsova, E.A.; Shpen, V.M. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertens. Dallas Tex 1979 1998, 31, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Orlov, S.N.; Klimanova, E.A.; Tverskoi, A.M.; Vladychenskaya, E.A.; Smolyaninova, L.V.; Lopina, O.D. Na+i, K+i-Dependent and-Independent Signaling Triggered by Cardiotonic Steroids: Facts and Artifacts. Molecules 2017, 22, 635. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-J.; Liu, W.; Yin, M. Trans fatty acids enhanced β-amyloid induced oxidative stress in nerve growth factor differentiated PC12 cells. Neurochem. Res. 2012, 37, 786–794. [Google Scholar] [CrossRef]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Ding, Y.; Zhao, J.; Zhang, X.; Wang, S.; Viola, K.L.; Chow, F.E.; Zhang, Y.; Lippa, C.; Klein, W.L.; Gong, Y. Amyloid Beta Oligomers Target to Extracellular and Intracellular Neuronal Synaptic Proteins in Alzheimer’s Disease. Front. Neurol. 2019, 10, 1140. [Google Scholar] [CrossRef]
- Jarosz-Griffiths, H.H.; Noble, E.; Rushworth, J.V.; Hooper, N.M. Amyloid-β Receptors: The Good, the Bad, and the Prion Protein. J. Biol. Chem. 2016, 291, 3174–3183. [Google Scholar] [CrossRef]
- Madan, N.; Xu, Y.; Duan, Q.; Banerjee, M.; Larre, I.; Pierre, S.V.; Xie, Z. Src-independent ERK signaling through the rat α3 isoform of Na/K-ATPase. Am. J. Physiol. Cell Physiol. 2017, 312, C222–C232. [Google Scholar] [CrossRef]
- Karpova, L.; Eva, A.; Kirch, U.; Boldyrev, A.; Scheiner-Bobis, G. Sodium pump alpha1 and alpha3 subunit isoforms mediate distinct responses to ouabain and are both essential for survival of human neuroblastoma. FEBS J. 2010, 277, 1853–1860. [Google Scholar] [CrossRef]
- Yosef, E.; Katz, A.; Peleg, Y.; Mehlman, T.; Karlish, S.J.D. Do Src Kinase and Caveolin Interact Directly with Na,K-ATPase? J. Biol. Chem. 2016, 291, 11736–11750. [Google Scholar] [CrossRef] [PubMed]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Li, X.; Liang, M.; Liu, L.; Xie, J.X.; Ye, Q.; Kometiani, P.; Tillekeratne, M.; Jin, R.; Xie, Z. Changes in Sodium Pump Expression Dictate the Effects of Ouabain on Cell Growth. J Biol Chem 2009, 284, 14921–14929. [Google Scholar] [CrossRef] [PubMed]
- Bae, O.-N.; Rajanikant, K.; Min, J.; Smith, J.; Baek, S.-H.; Serfozo, K.; Hejabian, S.; Lee, K.Y.; Kassab, M.; Majid, A. Lymphocyte cell kinase activation mediates neuroprotection during ischemic preconditioning. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 7278–7286. [Google Scholar] [CrossRef]
- Hossain, M.I.; Hoque, A.; Lessene, G.; Aizuddin Kamaruddin, M.; Chu, P.W.Y.; Ng, I.H.W.; Irtegun, S.; Ng, D.C.H.; Bogoyevitch, M.A.; Burgess, A.W.; et al. Dual role of Src kinase in governing neuronal survival. Brain Res. 2015, 1594, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Maness, P.F. Nonreceptor protein tyrosine kinases associated with neuronal development. Dev. Neurosci. 1992, 14, 257–270. [Google Scholar] [CrossRef]
- Dunning, C.J.R.; Black, H.L.; Andrews, K.L.; Davenport, E.C.; Conboy, M.; Chawla, S.; Dowle, A.A.; Ashford, D.; Thomas, J.R.; Evans, G.J.O. Multisite tyrosine phosphorylation of the N-terminus of Mint1/X11α by Src kinase regulates the trafficking of amyloid precursor protein. J. Neurochem. 2016, 137, 518–527. [Google Scholar] [CrossRef]
- Pimenova, A.A.; Thathiah, A.; De Strooper, B.; Tesseur, I. Regulation of amyloid precursor protein processing by serotonin signaling. PLoS ONE 2014, 9, e87014. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Hypoxia/ischemia activate processing of Amyloid Precursor Protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J. Neurochem. 2017, 140, 536–549. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; D’Souza, G.X.; Shi, J.; Ritter, A.; Suazo, J.; Sabbagh, M.N. The Cause of Alzheimer’s Disease: The Theory of Multipathology Convergence to Chronic Neuronal Stress. Aging Dis. 2022, 13, 37–60. [Google Scholar] [CrossRef]
- Yue, X.; Zhou, Y.; Qiao, M.; Zhao, X.; Huang, X.; Zhao, T.; Cheng, X.; Fan, M.; Zhao, Y.; Chen, R.; et al. Intermittent hypoxia treatment alleviates memory impairment in the 6-month-old APPswe/PS1dE9 mice and reduces amyloid beta accumulation and inflammation in the brain. Alzheimers Res. Ther. 2021, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic α subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Tolstova, A.P.; Strelkova, M.A.; Mitkevich, V.A.; Petrushanko, I.Y.; Makarov, A.A. Interaction Interface of Aβ42 with Human Na,K-ATPase Studied by MD and ITC and Inhibitor Screening by MD. Biomedicines 2022, 10, 1663. [Google Scholar] [CrossRef]
- Boldyrev, A.; Bulygina, E.; Yuneva, M.; Schoner, W. Na/K-ATPase regulates intracellular ROS level in cerebellum neurons. Ann. N. Y. Acad. Sci. 2003, 986, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.-J.; Abraham, N.G.; et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef]
- Yan, X.; Xun, M.; Li, J.; Wu, L.; Dou, X.; Zheng, J. Activation of Na+/K+-ATPase attenuates high glucose-induced H9c2 cell apoptosis via suppressing ROS accumulation and MAPKs activities by DRm217. Acta Biochim. Biophys. Sin. 2016, 48, 883–893. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef]
- Lakunina, V.A.; Petrushanko, I.Y.; Burnysheva, K.M.; Mitkevich, V.A.; Makarov, A.A. Alzheimer’s disease Aβ42 peptide induces an increase in Na,K-ATPase glutathionylation. Dokl. Biochem. Biophys. 2017, 473, 114–117. [Google Scholar] [CrossRef]
- Dyer, R.R.; Ford, K.I.; Robinson, R.A.S. The roles of S-nitrosylation and S-glutathionylation in Alzheimer’s disease. Methods Enzymol. 2019, 626, 499–538. [Google Scholar] [CrossRef]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef]
- Pocernich, C.B.; Lange, M.L.B.; Sultana, R.; Butterfield, D.A. Nutritional approaches to modulate oxidative stress in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 452–469. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Bader Lange, M.L.; Sultana, R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Markesbery, W.R. Oxidative damage in mild cognitive impairment and early Alzheimer’s disease. J. Neurosci. Res. 2007, 85, 3036–3040. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrushanko, I.Y.; Tverskoi, A.M.; Barykin, E.P.; Petrovskaya, A.V.; Strelkova, M.A.; Leonova, O.G.; Anashkina, A.A.; Tolstova, A.P.; Adzhubei, A.A.; Bogdanova, A.Y.; et al. Na,K-ATPase Acts as a Beta-Amyloid Receptor Triggering Src Kinase Activation. Cells 2022, 11, 2753. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172753
Petrushanko IY, Tverskoi AM, Barykin EP, Petrovskaya AV, Strelkova MA, Leonova OG, Anashkina AA, Tolstova AP, Adzhubei AA, Bogdanova AY, et al. Na,K-ATPase Acts as a Beta-Amyloid Receptor Triggering Src Kinase Activation. Cells. 2022; 11(17):2753. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172753
Chicago/Turabian StylePetrushanko, Irina Yu., Artem M. Tverskoi, Evgeny P. Barykin, Aleksandra V. Petrovskaya, Maria A. Strelkova, Olga G. Leonova, Anastasia A. Anashkina, Anna P. Tolstova, Alexei A. Adzhubei, Anna Yu. Bogdanova, and et al. 2022. "Na,K-ATPase Acts as a Beta-Amyloid Receptor Triggering Src Kinase Activation" Cells 11, no. 17: 2753. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172753