

Novel GATA1 Variant Causing a Bleeding Phenotype Associated with Combined Platelet α-/δ-Storage Pool Deficiency and Mild Dyserythropoiesis Modified by a SLC4A1 Variant
 , , and
, , and
Abstract
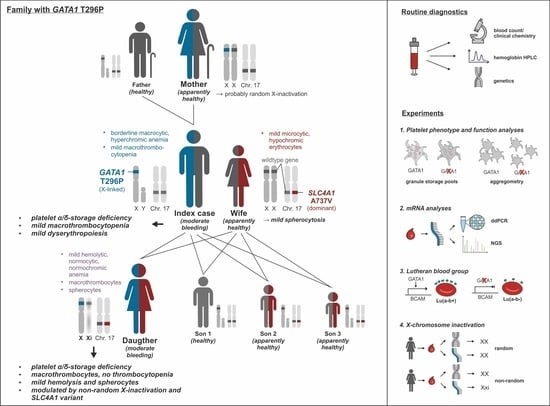
:
1. Introduction
2. Materials and Methods
2.1. Index Patient and Family Members
2.2. Blood Count, Clinical Chemistry, and HPLC
2.3. Whole Exome Sequencing
2.4. X-Chromosome Inactivation
2.5. Lutheran Blood Group Typing
2.6. Preparation of Platelet-Rich and Platelet-Free Plasma
2.7. Preparation of Washed Platelets for Electron Microscopy
2.8. Preparation of Gel-Filtered Platelets for Immunoblot Analysis and ELISA Assays
2.9. Light Transmission Aggregometry
2.10. Lumi-Aggregometry
2.11. Flow Cytometric Analysis of Platelet Receptor Surface Expression
2.12. Flow Cytometric Analysis of Platelet Functions
2.13. Transmission Electron Microscopy
2.14. Immunoblot Analysis
2.15. Quantification of Serotonin of Gel-Filtered Platelets
2.16. mRNA Profiling
2.17. Transcript Analysis via Digital Droplet PCR (ddPCR)
3. Results
3.1. Complete Blood Count, and Platelet and Erythrocyte Characteristics
3.2. Molecular Genetic Testing
3.3. Classification of the Candidate Variants Potentially Causing a Platelet Disorder
3.4. Classification of the SLC4A1 Variant Causitive for Mild Spherocytosis
3.5. Platelets from Index Patient and His Daughter Showed Impaired Aggregation in Response to ADP, Epinephrine, TRAP-6, and TxA2 Mimetic U46619, but Normal Surface Expression of Major Receptors
3.6. The Index Patient and His Daughter Presented with a Platelet δ-Granule ATP Secretion Defect When Platelet Aggregation Was Nearly Normally Induced by High Concentrations of Collagen and Arachidonic Acid
3.7. GATA1 T296P Variant Was Associated with a Combined Platelet α-/δ-Storage Pool Deficiency, Characterized with Reduced Numbers of α- and δ-Granules, Where Residual δ-Granules Lack ADP/ATP but Not Serotonin
3.8. mRNA Profiling of GATA1-Regulated Genes in Whole Blood from the Index, His Wife, and His Daughter
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Balduini, C.L.M.F.; Pecci, A. Inherited thrombocytopenias. In Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics: An Update; Gresele, P., Kleiman, N.S., Lopez, J.A., Page, C.P., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 727–747. [Google Scholar]
- Palma-Barqueros, V.; Revilla, N.; Sanchez, A.; Zamora Canovas, A.; Rodriguez-Alen, A.; Marin-Quilez, A.; Gonzalez-Porras, J.R.; Vicente, V.; Lozano, M.L.; Bastida, J.M.; et al. Inherited Platelet Disorders: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 4521. [Google Scholar] [CrossRef]
- Bury, L.; Falcinelli, E.; Gresele, P. Learning the Ropes of Platelet Count Regulation: Inherited Thrombocytopenias. J. Clin. Med. 2021, 10, 533. [Google Scholar] [CrossRef]
- Jurk, K.; Shiravand, Y. Platelet Phenotyping and Function Testing in Thrombocytopenia. J. Clin. Med. 2021, 10, 1114. [Google Scholar] [CrossRef]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the Platelet Physiology SSC. J. Thromb. Haemost. JTH 2020, 18, 732–739. [Google Scholar] [CrossRef]
- Songdej, N.; Rao, A.K. Hematopoietic transcription factor mutations: Important players in inherited platelet defects. Blood 2017, 129, 2873–2881. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.E. Transcription factor defects causing platelet disorders. Blood Rev. 2017, 31, 1–10. [Google Scholar] [CrossRef]
- Crispino, J.D.; Horwitz, M.S. GATA factor mutations in hematologic disease. Blood 2017, 129, 2103–2110. [Google Scholar] [CrossRef]
- Freson, K.; Wijgaerts, A.; Van Geet, C. GATA1 gene variants associated with thrombocytopenia and anemia. Platelets 2017, 28, 731–734. [Google Scholar] [CrossRef]
- Balduini, C.L.; De Candia, E.; Savoia, A. Why the disorder induced by GATA1 Arg216Gln mutation should be called “X-linked thrombocytopenia with thalassemia” rather than “X-linked gray platelet syndrome”. Blood 2007, 110, 2770–2771, author reply 2771. [Google Scholar] [CrossRef] [PubMed]
- Nurden, A.T.; Nurden, P. Should any genetic defect affecting alpha-granules in platelets be classified as gray platelet syndrome? Am. J. Hematol. 2016, 91, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, L.S.; Lareau, C.A.; Bao, E.L.; Liu, N.; Utsugisawa, T.; Tseng, A.M.; Myers, S.A.; Verboon, J.M.; Ulirsch, J.C.; Luo, W.; et al. Congenital anemia reveals distinct targeting mechanisms for master transcription factor GATA1. Blood 2022, 139, 2534–2546. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, H.; Buchler, E.; Wenzel, J.J.; Neukirch, C.; du Prel, J.B.; Lackner, K.J. Evaluation of a new pooling strategy based on leukocyte count for rapid quantification of allele frequencies. Clin. Chem. 2007, 53, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Bickmann, J.K.; Kamin, W.; Wiebel, M.; Hauser, F.; Wenzel, J.J.; Neukirch, C.; Stuhrmann, M.; Lackner, K.J.; Rossmann, H. A novel approach to CFTR mutation testing by pyrosequencing-based assay panels adapted to ethnicities. Clin. Chem. 2009, 55, 1083–1091. [Google Scholar] [CrossRef]
- Lahav, J.; Jurk, K.; Hess, O.; Barnes, M.J.; Farndale, R.W.; Luboshitz, J.; Kehrel, B.E. Sustained integrin ligation involves extracellular free sulfhydryls and enzymatically catalyzed disulfide exchange. Blood 2002, 100, 2472–2478. [Google Scholar] [CrossRef]
- Jurk, K.; Lahav, J.; van Aken, H.; Brodde, M.F.; Nofer, J.R.; Kehrel, B.E. Extracellular protein disulfide isomerase regulates feedback activation of platelet thrombin generation via modulation of coagulation factor binding. J. Thromb. Haemost. JTH 2011, 9, 2278–2290. [Google Scholar] [CrossRef]
- Kumm, E.J.; Pagel, O.; Gambaryan, S.; Walter, U.; Zahedi, R.P.; Smolenski, A.; Jurk, K. The Cell Cycle Checkpoint System MAST(L)-ENSA/ARPP19-PP2A is Targeted by cAMP/PKA and cGMP/PKG in Anucleate Human Platelets. Cells 2020, 9, 472. [Google Scholar] [CrossRef]
- Born, G.V. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- Loroch, S.; Trabold, K.; Gambaryan, S.; Reiss, C.; Schwierczek, K.; Fleming, I.; Sickmann, A.; Behnisch, W.; Zieger, B.; Zahedi, R.P.; et al. Alterations of the platelet proteome in type I Glanzmann thrombasthenia caused by different homozygous delG frameshift mutations in ITGA2B. Thromb. Haemost. 2017, 117, 556–569. [Google Scholar] [CrossRef]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J. Thromb. Haemost. JTH 2013, 11, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, A.; Bordet, J.C.; Eckly, A.; Gachet, C. Platelet delta-Storage Pool Disease: An Update. J. Clin. Med. 2020, 9, 2508. [Google Scholar] [CrossRef] [PubMed]
- Dohrmann, M.; Makhoul, S.; Gross, K.; Krause, M.; Pillitteri, D.; von Auer, C.; Walter, U.; Lutz, J.; Volf, I.; Kehrel, B.E.; et al. CD36-fibrin interaction propagates FXI-dependent thrombin generation of human platelets. FASEB J. 2020, 34, 9337–9357. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Lentjes, M.H.; Niessen, H.E.; Akiyama, Y.; de Bruine, A.P.; Melotte, V.; van Engeland, M. The emerging role of GATA transcription factors in development and disease. Expert Rev. Mol. Med. 2016, 18, e3. [Google Scholar] [CrossRef]
- Ling, T.; Crispino, J.D. GATA1 mutations in red cell disorders. IUBMB Life 2020, 72, 106–118. [Google Scholar] [CrossRef]
- Pereira, J.; Bento, C.; Manco, L.; Gonzalez, A.; Vagace, J.; Ribeiro, M.L. Congenital dyserythropoietic anemia associated to a GATA1 mutation aggravated by pyruvate kinase deficiency. Ann. Hematol. 2016, 95, 1551–1553. [Google Scholar] [CrossRef]
- Bastida, J.M.; Boeckelmann, D.; Palma-Barqueros, V.; Wolter, M.; LLozano, M.; Glonnegger, H.; Benito, R.; HSchilling, F.; Morgan, N.; Freson, K.; et al. A Novel Hemizygous Variant in GATA1 Associated with Bleeding Diathesis and Platelet Dysfunction in Two Unrelated Patients. Res. Pr. Thromb. Haemost. 2021, 5, PB0896. [Google Scholar]
- Singleton, B.K.; Frayne, J.; Anstee, D.J. Blood group phenotypes resulting from mutations in erythroid transcription factors. Curr. Opin. Hematol. 2012, 19, 486–493. [Google Scholar] [CrossRef]
- Eyler, C.E.; Telen, M.J. The Lutheran glycoprotein: A multifunctional adhesion receptor. Transfusion 2006, 46, 668–677. [Google Scholar] [CrossRef]
- Singleton, B.K.; Burton, N.M.; Green, C.; Brady, R.L.; Anstee, D.J. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood 2008, 112, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Helias, V.; Saison, C.; Peyrard, T.; Vera, E.; Prehu, C.; Cartron, J.P.; Arnaud, L. Molecular analysis of the rare in(Lu) blood type: Toward decoding the phenotypic outcome of haploinsufficiency for the transcription factor KLF1. Hum. Mutat. 2013, 34, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Barbarani, G.; Fugazza, C.; Strouboulis, J.; Ronchi, A.E. The Pleiotropic Effects of GATA1 and KLF1 in Physiological Erythropoiesis and in Dyserythropoietic Disorders. Front. Physiol. 2019, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Prevost, N.; Woulfe, D.S.; Jiang, H.; Stalker, T.J.; Marchese, P.; Ruggeri, Z.M.; Brass, L.F. Eph kinases and ephrins support thrombus growth and stability by regulating integrin outside-in signaling in platelets. Proc. Natl. Acad. Sci. USA 2005, 102, 9820–9825. [Google Scholar] [CrossRef] [PubMed]
- Reithmeier, R.A.; Casey, J.R.; Kalli, A.C.; Sansom, M.S.; Alguel, Y.; Iwata, S. Band 3, the human red cell chloride/bicarbonate anion exchanger (AE1, SLC4A1), in a structural context. Biochim. Biophys Acta 2016, 1858, 1507–1532. [Google Scholar] [CrossRef] [PubMed]
- White, J.G.; Thomas, A. Platelet structural pathology in a patient with the X-linked GATA-1, R216Q mutation. Platelets 2009, 20, 41–49. [Google Scholar] [CrossRef]
- Klar, J.; Khalfallah, A.; Arzoo, P.S.; Gazda, H.T.; Dahl, N. Recurrent GATA1 mutations in Diamond-Blackfan anaemia. Br. J. Haematol. 2014, 166, 949–951. [Google Scholar] [CrossRef]
- Hollanda, L.M.; Lima, C.S.; Cunha, A.F.; Albuquerque, D.M.; Vassallo, J.; Ozelo, M.C.; Joazeiro, P.P.; Saad, S.T.; Costa, F.F. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat. Genet. 2006, 38, 807–812. [Google Scholar] [CrossRef]
- Singleton, B.K.; Roxby, D.J.; Stirling, J.W.; Spring, F.A.; Wilson, C.; Poole, J.; Anstee, D.J. A novel GATA1 mutation (Stop414Arg) in a family with the rare X-linked blood group Lu(a-b-) phenotype and mild macrothrombocytic thrombocytopenia. Br. J. Haematol. 2013, 161, 139–142. [Google Scholar] [CrossRef]
- Abdulhay, N.J.; Fiorini, C.; Verboon, J.M.; Ludwig, L.S.; Ulirsch, J.C.; Zieger, B.; Lareau, C.A.; Mi, X.; Roy, A.; Obeng, E.A.; et al. Impaired human hematopoiesis due to a cryptic intronic GATA1 splicing mutation. J. Exp. Med. 2019, 216, 1050–1060. [Google Scholar] [CrossRef]
- Saultier, P.; Cabantous, S.; Puceat, M.; Peiretti, F.; Bigot, T.; Saut, N.; Bordet, J.C.; Canault, M.; van Agthoven, J.; Loosveld, M.; et al. GATA1 pathogenic variants disrupt MYH10 silencing during megakaryopoiesis. J. Thromb. Haemost. JTH 2021, 19, 2287–2301. [Google Scholar] [CrossRef] [PubMed]
- Jedlitschky, G.; Cattaneo, M.; Lubenow, L.E.; Rosskopf, D.; Lecchi, A.; Artoni, A.; Motta, G.; Niessen, J.; Kroemer, H.K.; Greinacher, A. Role of MRP4 (ABCC4) in platelet adenine nucleotide-storage: Evidence from patients with delta-storage pool deficiencies. Am. J. Pathol. 2010, 176, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Jurk, K.; Jahn, U.R.; Van Aken, H.; Schriek, C.; Droste, D.W.; Ritter, M.A.; Bernd Ringelstein, E.; Kehrel, B.E. Platelets in patients with acute ischemic stroke are exhausted and refractory to thrombin, due to cleavage of the seven-transmembrane thrombin receptor (PAR-1). Thromb. Haemost. 2004, 91, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, Q.; Shang, X.; Zou, S.; Qin, J.; Li, K.; Lin, B.; Tao, Z.; Long, X.; Xu, X. Diamond-Blackfan anaemia caused by a de novo initiation codon mutation resulting in a shorter isoform of GATA1. Clin. Genet. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Hetzer, B.; Meryk, A.; Kropshofer, G.; Bargehr, C.; Jimenez-Heredia, R.; Boztug, K.; Muhlegger, B.E.; Dworzak, M.; Gruber, T.; Crazzolara, R. An R307H substitution in GATA1 that prevents Ser310 phosphorylation causes severe fetal anemia. Blood Adv. 2022, 6, 4330–4334. [Google Scholar] [CrossRef]
- Strouboulis, J.; Ronchi, A.E. GATA1: Function through disorder. Blood 2022, 139, 2422–2423. [Google Scholar] [CrossRef]
- Chou, S.T.; Kacena, M.A.; Weiss, M.J.; Raskind, W.H. GATA1-Related X-Linked Cytopenia. In GeneReviews((R)); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Van Dooijeweert, B.; Kia, S.K.; Dahl, N.; Fenneteau, O.; Leguit, R.; Nieuwenhuis, E.; van Solinge, W.; van Wijk, R.; Da Costa, L.; Bartels, M. GATA-1 Defects in Diamond-Blackfan Anemia: Phenotypic Characterization Points to a Specific Subset of Disease. Genes 2022, 13, 447. [Google Scholar] [CrossRef]
- Freson, K.; Devriendt, K.; Matthijs, G.; Van Hoof, A.; De Vos, R.; Thys, C.; Minner, K.; Hoylaerts, M.F.; Vermylen, J.; Van Geet, C. Platelet characteristics in patients with X-linked macrothrombocytopenia because of a novel GATA1 mutation. Blood 2001, 98, 85–92. [Google Scholar] [CrossRef]
- Freson, K.; Matthijs, G.; Thys, C.; Marien, P.; Hoylaerts, M.F.; Vermylen, J.; Van Geet, C. Different substitutions at residue D218 of the X-linked transcription factor GATA1 lead to altered clinical severity of macrothrombocytopenia and anemia and are associated with variable skewed X inactivation. Hum. Mol. Genet. 2002, 11, 147–152. [Google Scholar] [CrossRef]
- Hughan, S.C.; Senis, Y.; Best, D.; Thomas, A.; Frampton, J.; Vyas, P.; Watson, S.P. Selective impairment of platelet activation to collagen in the absence of GATA1. Blood 2005, 105, 4369–4376. [Google Scholar] [CrossRef]
- Astrom, M.; Hahn-Stromberg, V.; Zetterberg, E.; Vedin, I.; Merup, M.; Palmblad, J. X-linked thrombocytopenia with thalassemia displays bone marrow reticulin fibrosis and enhanced angiogenesis: Comparisons with primary myelofibrosis. Am. J. Hematol. 2015, 90, E44–E48. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana, M.; Antoniani, C.; Miccio, A. Gene Therapy for beta-Hemoglobinopathies. Mol. Ther. 2017, 25, 1142–1154. [Google Scholar] [CrossRef] [PubMed]
- White, J.G. Platelet pathology in carriers of the X-linked GATA-1 macrothrombocytopenia. Platelets 2007, 18, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.D.; Steensma, D.P.; Pulsipher, M.A.; Spangrude, G.J.; Kushner, J.P. Congenital erythropoietic porphyria due to a mutation in GATA1: The first trans-acting mutation causative for a human porphyria. Blood 2007, 109, 2618–2621. [Google Scholar] [CrossRef] [Green Version]
- Doshi, B.S.; Abramowsky, C.; Briones, M.; Bunting, S.T. Concomitant a novel ALAS2 mutation and GATA1 mutation in a newborn: A case report and review of the literature. Am. J. Blood Res. 2014, 4, 41–45. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Index | Daughter | Wife | Reference | Unit | |
|---|---|---|---|---|---|---|
| White blood cell count | 7.5 | 6.80 (5.5–15.5) | 6.20 | 3.5–10 | ×103/µL | |
| Automated differential blood count | normal | normal | normal | normal | - | |
| Red blood cell count | −3.70 (4.3–6.3) | 3.63 (3.1–5.2) | 4.07 (3.7–4.8) | indiv. | ×106/µL | |
| Platelets | −88 | 178.0 | 273.0 | 150–360 | ×103/µL | |
| Mean platelet volume | 10.4 | 11.2 | 9.9 | 7.6–11.2 | fL | |
| Hemoglobin | −12.5 (13.5–17.5) | 10.9 (10–13) | −11.6 (12–16) | indiv. | g/dL | |
| Hemoglobin HPLC | HbF | +2.8 | ++13.5 | +1.8 | <1 | % |
| HbA2 | 3.0 | 2.2 | 2.7 | <3.3 (β-thalassemia) | % | |
| Variant hemoglobin | none | none | none | none | - | |
| Hematocrit | −37.2 (39–49) | 31.8 (30–45) | (−)33.5 (34–44) | indiv. | % | |
| Mean corpuscular volume | (+)100.3 | 87.6 | (−)82.3 | 83–100 | fL | |
| Mean corpuscular hemoglobin | (+)33.7 | 30.0 | 28.5 | 27–33 | pg | |
| MCHC | 33.6 | 34.3 | 34.6 | 32–35 | g/dL | |
| RDW-CV | 14.3 | 13.9 | 14.2 | 11–15 | % | |
| Blood smear | White blood cells | normal | normal | normal | normal | - |
| Red blood cells | normal | + spherocytes | ++ spherocytes | normal | - | |
| Platelets | macrothrombocytes | macrothrombocytes | normal | normal | - | |
| C-reactive protein | 2.2 | 0.20 | n.d. | <5 | mg/L | |
| Ferritin | +332 (20–275) | +35 (6–24) | 9.4 (5–200) | indiv. | ng/mL | |
| Transferrin saturation | −15.1 | 38.9 (7–44) | −−7.4 | 16–45 | % | |
| Haptoglobin | 0.84 (0.14–2.58) | −<0.08 (0.11–2.2) | 1.69 (0.35–2.50) | indiv. | g/L | |
| Zinc protoporphyrin | 75 | 80 | (+)104 | >100 (iron deficiency) | mmol/mol heme | |
| Reticulocytes abs. | +210 | +114 | +78 | 19–69 | /nL | |
| Erythropoietin | 19.1 | +37.1 | 13.2 | 4.3–29 | mU/mL | |
| Lutheran blood group | a-b- | a-b+ | a-b+ | a-b+ | - | |
| GATA1 | EPHB1 | AP3B1 | SLC4A1 | ||
|---|---|---|---|---|---|
| c.886A>C | c.1856T>C | c.1069A>G | c.2210C>T | ||
| p.(Thr296Pro) | p.(Val619Ala) | p.(Ile357Val) | p.(Ala737Val) | ||
| Reference sequence | NM_002049.4 | NM_004441.5 | NM_003664.5 | NM_000342.4 | |
| Chromosome | X | 3 | 5 | 17 | |
| Exon | 6 | 10 | 10 | 17 | |
| dbSNP | no entry | rs375222902 | rs142025324 | rs886052997 | |
| ClinVar (number of entries) | no entry | no entry | VUS (2), likely benign (4) | VUS (3) | |
| MAF in GnomAD | no entry | 0.0001532 | 0.001795 | 0.000003982 | |
| Mutation Taster | disease causing | disease causing | disease causing | disease causing | |
| PolyPhen2 | probably damaging | probably damaging | benign | probably damaging | |
| SIFT | deleterious | deleterious | tolerated | deleterious | |
| PROVEAN | deleterious | deleterious | neutral | deleterious | |
| OMIM Gene | 305371 | 600600 | 603401 | 109270 | |
| OMIM Disease | 300367 (XLTDA) | no entry | 608233 (HPS2) | 612653 (SPH4) | |
| Inheritance (OMIM) | XLR | no entry | AR | AD | |
| ACMG criteria | pathogenic | VUS | likely benign | pathogenic | |
| Variant-related symptoms in the affected family members | macrothrombocytopenia, mild dyserythropoesis | none | none | mild spherocytosis, potential modification of GATA1 disease | |
| NGS result confirmed | yes (pyrosequencing) | yes (Sanger sequencing) | yes (Sanger sequencing) | yes (Sanger sequencing) | |
| Part of in-house coagulation panel (322 genes) | ✓ | ✓ | ✓ | - | |
| Part of in-house anemia panel (96 genes) | ✓ | - | - | ✓ | |
| Genotype | index (bleeding) | C | TC | AG | CC |
| wife (spherocytes) | AA | TT | AA | CT | |
| daughter (bleeding, spherocytes, hemolysis) | AC | TC | AG | CT | |
| son 1 (healthy) | A | TT | AA | CC | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurk, K.; Adenaeuer, A.; Sollfrank, S.; Groß, K.; Häuser, F.; Czwalinna, A.; Erkel, J.; Fritsch, N.; Marandiuc, D.; Schaller, M.; et al. Novel GATA1 Variant Causing a Bleeding Phenotype Associated with Combined Platelet α-/δ-Storage Pool Deficiency and Mild Dyserythropoiesis Modified by a SLC4A1 Variant. Cells 2022, 11, 3071. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11193071
Jurk K, Adenaeuer A, Sollfrank S, Groß K, Häuser F, Czwalinna A, Erkel J, Fritsch N, Marandiuc D, Schaller M, et al. Novel GATA1 Variant Causing a Bleeding Phenotype Associated with Combined Platelet α-/δ-Storage Pool Deficiency and Mild Dyserythropoiesis Modified by a SLC4A1 Variant. Cells. 2022; 11(19):3071. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11193071
Chicago/Turabian StyleJurk, Kerstin, Anke Adenaeuer, Stefanie Sollfrank, Kathrin Groß, Friederike Häuser, Andreas Czwalinna, Josef Erkel, Nele Fritsch, Dana Marandiuc, Martin Schaller, and et al. 2022. "Novel GATA1 Variant Causing a Bleeding Phenotype Associated with Combined Platelet α-/δ-Storage Pool Deficiency and Mild Dyserythropoiesis Modified by a SLC4A1 Variant" Cells 11, no. 19: 3071. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11193071