
Role of Bioactive Compounds in the Regulation of Mitochondrial Dysfunctions in Brain and Age-Related Neurodegenerative Diseases

 ,
,  , ,
, , 
 and
and 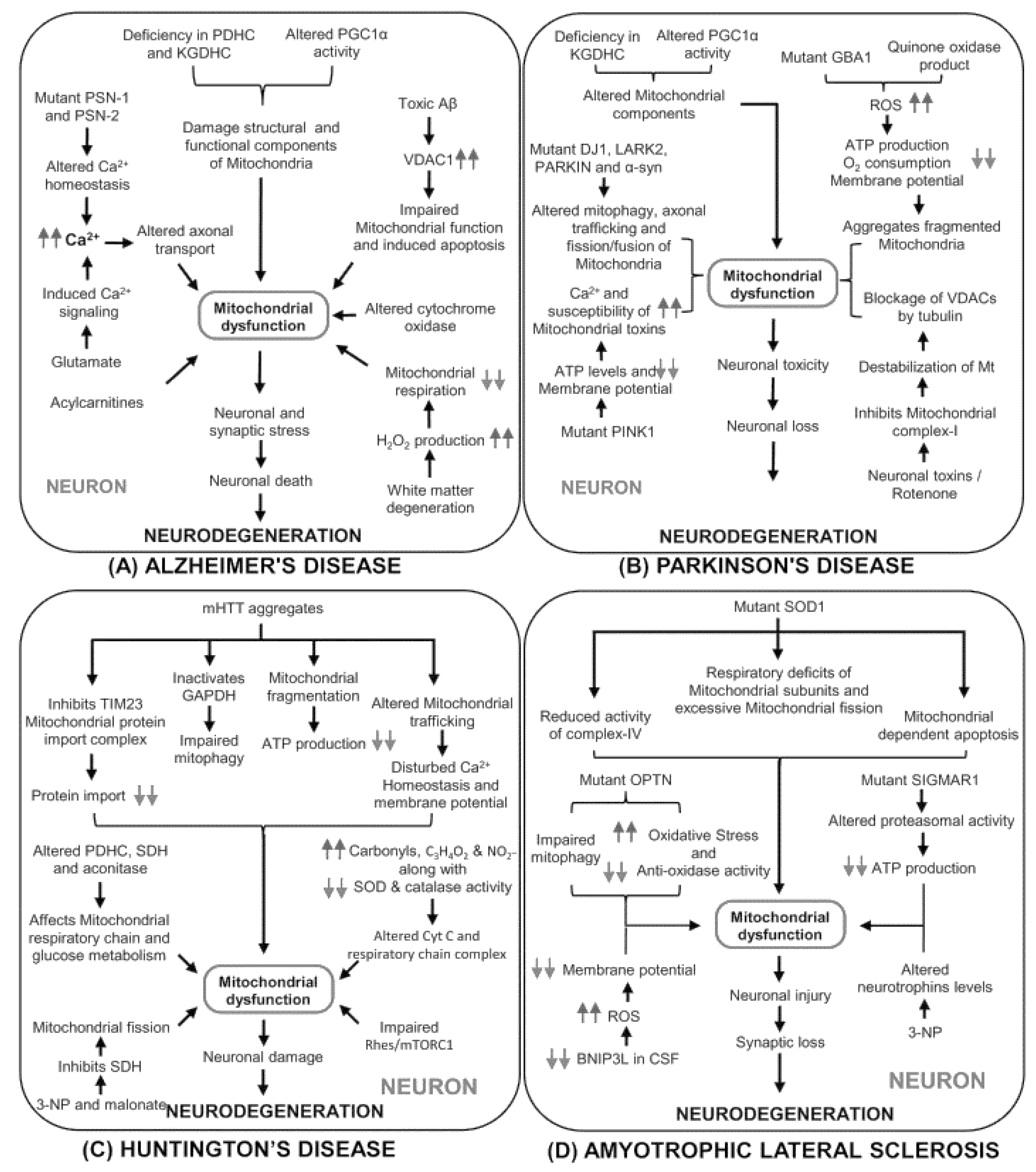{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Involvement of Mitochondria in Neuronal Functions
3. Mitochondrial and Neurodegenerative Disorder
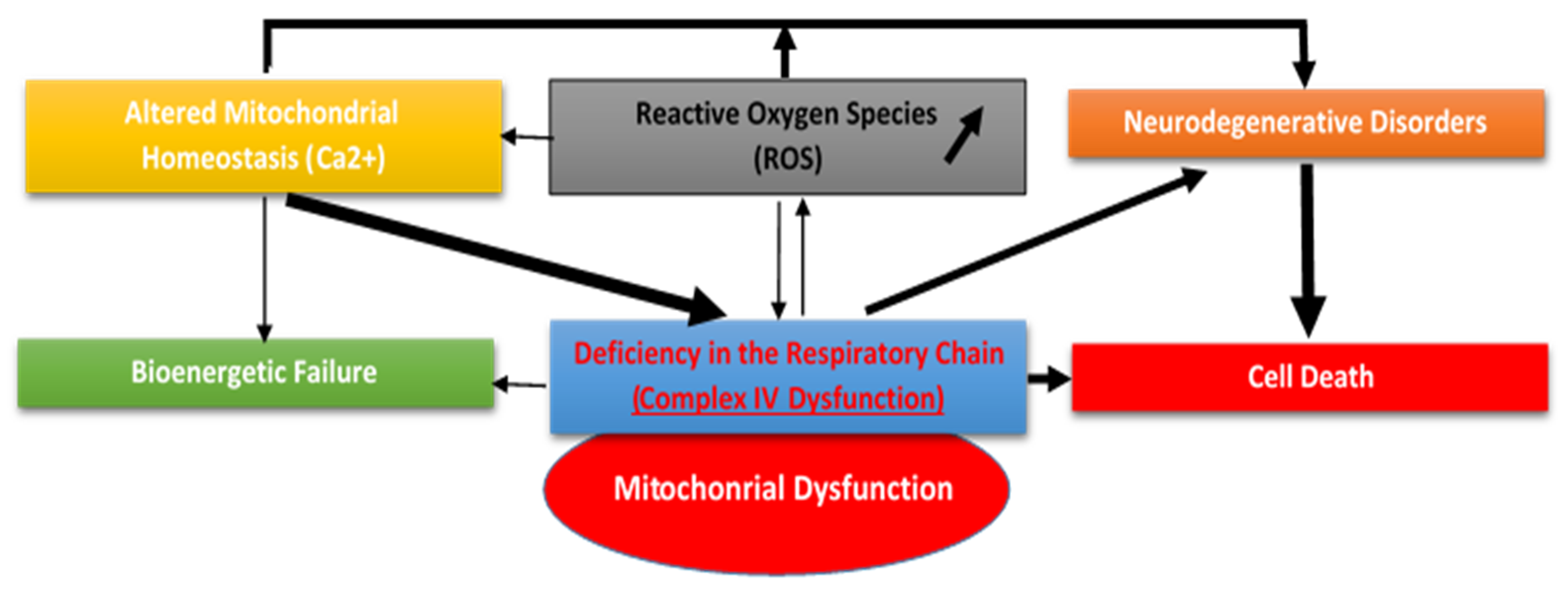
4. Mitochondrial Dysfunction and Oxidative Stress
5. Mitochondria and Apoptosis Mechanism
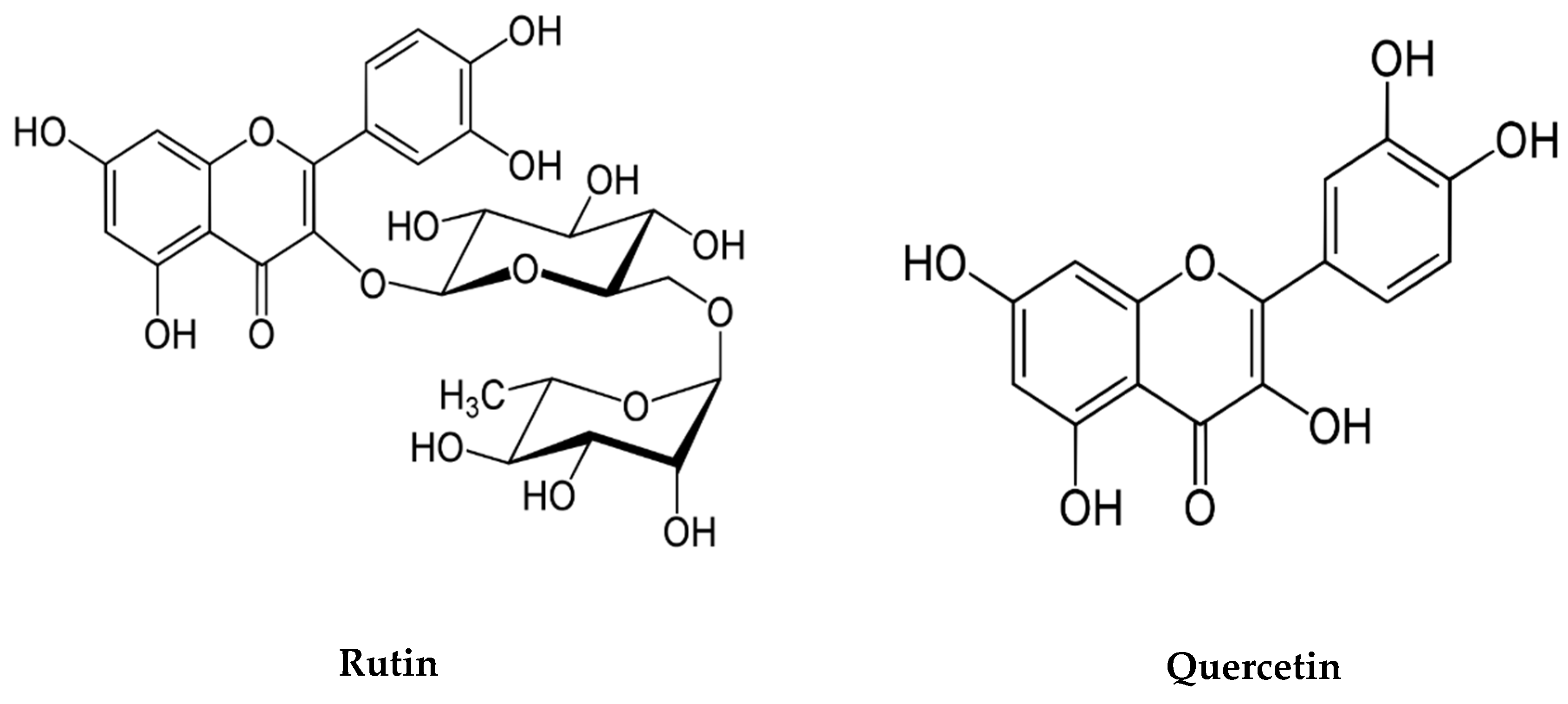
6. Neuroprotective Activities of Bioactive Compounds
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate, Glutamate Receptors, and Downstream Signaling Pathways. Int. J. Biol. Sci. 2013, 9, 948–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacobbo, B.L.; Doorduin, J.; Klein, C.H.; Dierckx, R.A.J.O.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karis, K.; Eskla, K.-L.; Kaare, M.; Täht, K.; Tuusov, J.; Visnapuu, T.; Innos, J.; Jayaram, M.; Timmusk, T.; Weickert, C.S.; et al. Altered Expression Profile of IgLON Family of Neural Cell Adhesion Molecules in the Dorsolateral Prefrontal Cortex of Schizophrenic Patients. Front. Mol. Neurosci. 2018, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.H.; Liang, Z.Q.; Gu, Z.L.; Yang, Y.P.; Reid, P.; Qin, Z.H. Contributions of autophagic and apoptotic mechanisms to CrTX-induced death of K562 cells. Toxicon 2006, 47, 521–530. [Google Scholar] [CrossRef]
- Bloss, E.B.; Cembrowski, M.S.; Karsh, B.; Colonell, J.; Fetter, R.D.; Spruston, N. Single excitatory axons form clustered synapses onto CA1 pyramidal cell dendrites. Nat. Neurosci. 2018, 21, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Alshaabi, H.; Heininger, M.; Cunniff, B. Dynamic regulation of subcellular mitochondrial position for localized metabolite levels. J. Biochem. 2020, 167, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef]
- Zhang, Y.; Williams, P.R.; Jacobi, A.; Wang, C.; Goe, A.; Hirano, A.A.; Brecha, N.C.; Kerschensteiner, D.; He, Z. Elevating Growth Factor Responsiveness and Axon Regeneration by Modulating Presynaptic Inputs. Neuron 2019, 103, 39–51. [Google Scholar] [CrossRef]
- Ledda, F.; Paratcha, G. Mechanisms regulating dendritic arbor patterning. Cell. Mol. Life Sci. 2017, 74, 4511–4537. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M. International Natural Product Sciences Taskforce, Supuran CT. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Sarangarajan, R.; Meera, S.; Rukkumani, R.; Sankar, P.; Anuradha, G. Antioxidants: Friend or foe? Asian Pac. J. Trop. Med. 2017, 10, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Lalkovičová, M.; Danielisová, V. Neuroprotection and antioxidants. Neural Regen. Res. 2016, 11, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Neha, K.; Haider, M.R.; Pathak, A.; Yar, M.S. Medicinal prospects of antioxidants: A review. Eur. J. Med. Chem. 2019, 178, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M.; Negut, I.; Grumezescu, V.; Grumezescu, A.M.; Teleanu, R.I. Nanomaterials for drug delivery to the central nervous system. Nanomaterials 2019, 9, 371. [Google Scholar] [CrossRef] [Green Version]
- Teleanu, D.M.; Chircov, C.; Grumezescu, A.M.; Teleanu, R.I. Neuronanomedicine: An up-to-date overview. Pharmaceutics 2019, 11, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benaroya, H. Brain energetics, mitochondria, and traumatic brain injury. Rev. Neurosci. 2020, 31, 363–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.H.H.; Chan, J.Y.H. Mitochondria and Reactive Oxygen Species Contribute to Neurogenic Hypertension. Physiology 2017, 32, 308–321. [Google Scholar] [CrossRef]
- Chandra, G.; Shenoi, R.A.; An, R.; Rajamma, U.; Mohanakumar, K.P. Reinforcing mitochondrial functions in aging brain: An insight into Parkinson’s disease therapeutics. J. Chem. Neuroanat. 2019, 95, 29–42. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [Green Version]
- Clausen, A.; McClanahan, T.; Ji, S.G.; Weiss, J.H. Mechanisms of Rapid Reactive Oxygen Species Generation in Response to Cytosolic Ca2+ or Zn2+ Loads in Cortical Neurons. PLoS ONE 2013, 8, e83347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 1, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Lee, A.; Hirabayashi, Y.; Kwon, S.-K.; Lewis, T.L., Jr.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- de Jong, A.P.H.; Fioravante, D. Translating neuronal activity at the synapse: Presynaptic calcium sensors in short-term plasticity. Front. Cell. Neurosci. 2014, 8, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Arias-Reyes, C.; Losantos-Ramos, K.; Gonzales, M.; Furrer, D.; Soliz, J. NADH-linked mitochondrial respiration in the developing mouse brain is sex-, age-and tissue-dependent. Respir. Physiol. Neurobiol. 2019, 266, 156–162. [Google Scholar] [CrossRef]
- Ge, S.; Sailor, K.A.; Ming, G.L.; Song, H. Synaptic integration and plasticity of new neurons in the adult hippocampus. J. Physiol. 2008, 586, 3759–3765. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Saitoh, Y.; Takashima, N.; Murayama, A.; Niibori, Y.; Ageta, H.; Sekiguchi, M.; Sugiyama, H.; Inokuchi, K. Adult neurogenesis modulates the hippocampus-dependent period of associative fear memory. Cell 2009, 139, 814–827. [Google Scholar] [CrossRef] [Green Version]
- Hamby, M.E.; Coskun, V.; Sun, Y.E. Transcriptional regulation of neuronal differentiation: The epigenetic layer of complexity. Biochim. Biophys. Acta 2008, 1779, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Cane, K.N.; Anderson, C.R. Generating diversity: Mechanisms regulating the differentiation of autonomic neuron phenotypes. Auton. Neurosci. 2009, 151, 17–29. [Google Scholar] [CrossRef]
- Vayssière, J.L.; Cordeau-Lossouarn, L.; Larcher, J.C.; Basseville, M.; Gros, F.; Croizat, B. Participation of the mitochondrial genome in the differentiation of neuroblastoma cells. Vitro Cell. Dev. Biol. 1992, 28A, 763–772. [Google Scholar] [CrossRef]
- Chan, S.L.; Liu, D.; Kyriazis, G.A.; Bagsiyao, P.; Ouyang, X.; Mattson, M.P. Mitochondrial uncoupling protein-4 regulates calcium homeostasis and sensitivity to store depletion-induced apoptosis in neural cells. J. Biol. Chem. 2006, 281, 37391–37403. [Google Scholar] [CrossRef] [Green Version]
- Martynoga, B.; Drechsel, D.; Guillemot, F. Molecular control of neurogenesis: A view from the mammalian cerebral cortex. Cold Spring Harb. Perspect. Biol. 2012, 4, a008359. [Google Scholar] [CrossRef]
- Arredondo, S.B.; Valenzuela-Bezanilla, D.; Mardones, M.D.; Varela-Nallar, L. Role of Wnt Signaling in Adult Hippocampal Neurogenesis in Health and Disease. Front. Cell Dev. Biol. 2020, 8, 860. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, M.; Widmann, C. Burning fat to keep your stem cells? The role of fatty acid oxidation in various tissue stem cells. Curr. Opin. Lipidol. 2018, 29, 426–427. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Bharat, V.; Wang, X. Surveillance and transport of mitochondria in neurons. Curr. Opin. Neurobiol. 2019, 57, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bénit, P.; Slama, A.; Cartault, F.; Giurgea, I.; Chretien, D.; Lebon, S.; Marsac, C.; Munnich, A.; Rötig, A.; Rustin, P. Mutant NDUFS3 subunit of mitochondrial complex I cause Leigh syndrome. J. Med. Genet. 2004, 41, 14–17. [Google Scholar] [CrossRef]
- Andreazza, A.C.; Shao, L.; Wang, J.F.; Young, L.T. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch. Gen. Psychiatry 2010, 67, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Baradaran, R.; Berrisford, J.M.; Minhas, G.S.; Sazanov, L.A. Crystal structure of the entire respiratory complex I. Nature 2013, 494, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, J.; Wong, L.-J.C.; Gorman, E.B. Mitochondrial respiratory chain complex II. In Mitochondrial Disorders Caused by Nuclear Genes; Wong, L.-J.C., Ed.; Springer: New York, NY, USA, 2013; pp. 203–218. [Google Scholar]
- Dröse, S. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre-and postconditioning. Biochim. Biophys. Acta 2013, 1827, 578–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef]
- Jain-Ghai, S.; Cameron, J.M.; Al Maawali, A.; Blaser, S.; Kay, N.M.; Robinson, B.; Raimanal, J. Complex II deficiency—A case report and review of the literature. Am. J. Hum. Genet. 2013, 161, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Gimenez-Roqueplo, A.P.; Mannelli, M.; Pacak, K. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr.-Relat. Cancer 2009, 16, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanova, K.H.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Free. Radic. Res. 2020, 25, 26–32. [Google Scholar]
- Borisov, V.B. Defects in mitochondrial respiratory complexes III and IV, and human pathologies. Mol. Asp. Med. 2002, 23, 385–412. [Google Scholar] [CrossRef]
- Lott, M.T.; Leipzig, T.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA variation and analysis using Mitomap and Mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1.23.21–1.23.26. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell. Biol. 2006, 26, 4872–4881. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Nury, T.; Vejux, A.; Zarrouk, A.; Caccia, C.; Debbabi, M.; Fromont, A.; Sghaier, R.; Moreau, T.; Lizard, G. Mitochondrial dysfunctions in 7-ketocholesterol-treated 158N oligodendrocytes without or with α-tocopherol: Impacts on the cellular profil of tricarboxylic cycle-associated organic acids, long chain saturated and unsaturated fatty acids, oxysterols, cholesterol and cholesterol precursors. J. Steroid Biochem. Mol. Biol. 2017, 169, 96–110. [Google Scholar]
- Li, X.; Yang, Y. Mitochondrial disorders associated with mitochondrial respiratory chain complex V deficiency. Zhongguo Dang Dai Er Ke Za Zhi 2013, 15, 596–600. [Google Scholar]
- Stroud, D.A.; Maher, M.J.; Lindau, C.; Vögtle, F.-N.; Frazier, A.E.; Surgenor, E.; Mountford, H.; Singh, A.P.; Bonas, M.; Oeljeklaus, S.; et al. COA6 is a mitochondrial complex IV assembly factor critical for biogenesis of mtDNA-encoded COX2. Hum. Mol. Genet. 2015, 24, 5404–5415. [Google Scholar] [CrossRef] [Green Version]
- Mourier, A.; Ruzzenente, B.; Brandt, T.; Kuhlbrandt, W.; Larsson, N.G. Loss of LRPPRC causes ATP synthase deficiency. Hum. Mol. Genet. 2013, 23, 2580–2592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zink, A.; Priller, J.; Prigione, A. Pluripotent Stem Cells for Uncovering the Role of Mitochondria in Human Brain Function and Dysfunction. J. Mol. Biol. 2018, 430, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Monzio Compagnoni, G.; Di Fonzo, A.; Corti, S.; Comi, G.P.; Bresolin, N.; Masliah, E. The Role of Mitochondria in Neurodegenerative Diseases: The Lesson from Alzheimer’s Disease and Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 2959–2980. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.; Deisseroth, K. CLARITY for mapping the nervous system. Nat. Methods 2013, 10, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.; Laude, A.; Lightowlers, R.; Morris, C.M.; Turnbull, D.M.; Lax, N.Z. Development of passive CLARITY and immunofluorescent labelling of multiple proteins in human cerebellum: Understanding mechanisms of neurodegeneration in mitochondrial disease. Sci. Rep. 2016, 6, 26013. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, H.; Goto, Y.I. Heteroplasmic mitochondrial DNA mutations and mitochondrial diseases: Toward iPSC-based disease modeling, drug discovery, and regenerative therapeutics. Stem Cells 2016, 34, 801–808. [Google Scholar] [CrossRef]
- Peralta, S.; Torraco, A.; Iommarini, L.; Diaz, F. Mitochondrial diseases Part III: Therapeutic interventions in mouse models of OXPHOS deficiencies. Mitochondrion 2015, 23, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Torraco, A.; Peralta, S.; Iommarini, L.; Diaz, F. Mitochondrial diseases Part I: Mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors. Mitochondrion 2015, 21, 76–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction-A Mini-Review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef] [Green Version]
- Hollensworth, S.B.; Shen, C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; LeDoux, S.P. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic. Biol. Med. 2000, 28, 1161–1174. [Google Scholar] [CrossRef]
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Voets, A.M.; Huigsloot, M.; Lindsey, P.J.; Leenders, A.M.; Koopman, W.J.; Willems, P.H.; Rodenburg, R.J.; Smeitink, J.A.; Smeets, H.J. Transcriptional changes in OXPHOS complex I deficiency are related to anti-oxidant pathways and could explain the disturbed calcium homeostasis. Biochim. Biophys. Acta 2012, 1822, 1161–1618. [Google Scholar] [CrossRef] [Green Version]
- Castro Mdel, R.; Suarez, E.; Kraiselburd, E.; Isidro, A.; Paz, J.; Ferder, L. Ayala-Torres, S. Aging increases mitochondrial DNA damage and oxidative stress in liver of rhesus monkeys. Exp. Gerontol. 2012, 47, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J. Neurol. Sci. 2007, 257, 221–239. [Google Scholar] [CrossRef]
- Ullah, H.; Di Minno, A.; Santarcangelo, C.; Khan, H.; Daglia, M. Improvement of Oxidative Stress and Mitochondrial Dysfunction by β-Caryophyllene: A Focus on the Nervous System. Antioxidants 2021, 10, 546. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H.; Lu, C.Y.; Wei, C.Y.; Ma, Y.S.; Lee, H.C. Oxidative stress in human aging and mitochondrial disease-consequences of defective mitochondrial respiration and impaired antioxidant enzyme system. Chin. J. Physiol. 2001, 44, 1–11. [Google Scholar]
- Selivanov, V.A.; Votyakova, T.V.; Pivtoraiko, V.N.; Zeak, J.; Sukhomlin, T.; Trucco, M.; Roca, J.; Cascante, M. Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput. Biol. 2011, 7, e1001115. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239–247. [Google Scholar] [CrossRef]
- Lin, M.T.; Cantuti-Castelvetri, I.; Zheng, K.; Jackson, K.E.; Tan, Y.B.; Arzberger, T.; Lees, A.J.; Betensky, R.A.; Bea, M.F.; Simon, D.K. Somatic mitochondrial DNA mutations in early Parkinson’s disease and incidental Lewy body disease. Ann. Neurol. 2012, 71, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapetanovic, R.; Bokil, N.J.; Sweet, M.J. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: A ticking time bomb in ageing. Ageing Res. Rev. 2015, 24, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Pedrini, S.; Caputo, L.; Annoni, G.; Davis, L.J.; Ferri, C.; Casadei, V.; Grimaldi, L.M. Increased plasma levels of interleukin-1, interleukin-6 and alpha-1-antichymotrypsin in patients with Alzheimer’s disease: Peripheral inflammation or signals from the brain? J. Neuroimmunol. 2000, 103, 97–102. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Bennett, J.P. An evaluation of the role of mitochondria in neurodegenerative diseases: Mitochondrial mutations and oxidative pathology, protective nuclear responses, and cell death in neurodegeneration. Brain Res. Brain Res. Rev. 1999, 29, 1–25. [Google Scholar] [CrossRef]
- Shevtsova, E.F.; Maltsev, A.A.; Vinogradova, D.V.; Shevtsov, P.N.; Bachurin, S.O. Mitochondria as a promising target for developing novel agents for treating Alzheimer’s disease. Med. Res. Rev. 2020, 41, 803–827. [Google Scholar] [CrossRef] [PubMed]
- Gulbins, E.; Dreschers, S.; Bock, J. Role of mitochondria in apoptosis. Exp. Physiol. 2003, 88, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. A Review on Ethnopharmacological Applications, Pharmacological Activities, and Bioactive Compounds of Mangifera indica (Mango). Evid.-Based Complement. Altern. Med. 2017, 2017, 6949835. [Google Scholar]
- Mohd Sairazi, N.S.; Sirajudeen, K.N.S. Natural Products and Their Bioactive Compounds: Neuroprotective Potentials against Neurodegenerative Diseases. Evid.-Based Complement. Altern. Med. 2020, 2020, 6565396. [Google Scholar] [CrossRef]
- Kharoubi, O.; Slimani, M.; Ait Hamadouche, N.; Krouf, D.; Aoues, A. Protective effect of Wormwood extract on lead induced neurotoxicity and cognitive disorded. Int. J. Green Pharm. 2010, 4, 193–198. [Google Scholar] [CrossRef]
- Bagheri, H.; Ghasemi, F.; Barreto, G.E.; Rafiee, R.; Sathyapalan, T.; Sahebkar, A. Effects of curcumin on mitochondria in neurodegenerative diseases. Biofactors 2020, 46, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Lahouel, Z.; Kharoubi, O.; Boussadia, A.; Bekkouche, Z.; Aoues, A. Effect of Aluminium and Aqueous extract of Rosmarinus officinalis on rat Brain: Impact on Neurobehavioral and Histological study. J. Drug Deliv. Ther. 2020, 10, 179–187. [Google Scholar] [CrossRef]
- Boussadia, A.; Kharoubi, O.; Lahouel, Z.; Benglia, A.; Aoues, A. Effect of aqueous Salvia officinalis extract on Aluminum chloride-induced neurotoxicity in female rats. Int. J. Pharm. Res. Allied Sci. 2020, 9, 139–150. [Google Scholar]
- Vuolo, M.M.; Lima, V.S.; Maróstica Junior, M.R. Chapter 2-phenolic compounds: Structure, classification, and antioxidant power. In Bioactive Compounds; Campos, M.R.S., Ed.; Woodhead Publishing: Cambridge, MA, USA, 2019; pp. 33–50. [Google Scholar]
- de la Rosa, L.A.; Moreno-Escamilla, J.O.; Rodrigo-García, J.; Alvarez-Parrilla, E. Chapter 12-phenolic compounds. In Postharvest Physiology and Biochemistry of Fruits and Vegetables; Yahia, E.M., Ed.; Woodhead Publishing: Cambridge, MA, USA, 2019; pp. 253–271. [Google Scholar]
- Minatel, I.O.; Borges, C.V.; Ferreira, M.I.; Gomez, H.A.G.; Chen, C.-Y.O.; Lima, G.P.P. Phenolic Compounds: Functional Properties, Impact of Processing and Bioavailability; Intech Open: London, UK, 2017; p. 236. [Google Scholar]
- Azam, S.; Jakaria, M.; Kim, I.-S.; Kim, J.; Haque, M.E.; Choi, D.-K. Regulation of toll-like receptor (tlr) signaling pathway by polyphenols in the treatment of age-linked neurodegenerative diseases: Focus on tlr4 signaling. Front. Immunol. 2019, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Frandsen, J.R.; Narayanasamy, P. Neuroprotection through flavonoid: Enhancement of the glyoxalase pathway. Redox Biol. 2019, 14, 465–473. [Google Scholar] [CrossRef]
- Šegota, S.; Crnolatac, I.; Čadež, V.; Jembrek, M.J.; Sikirić, M.D. Neuroprotection and neuronal recovery under the oxidative stress achieved by enhanced lipid membrane interaction with flavonoids. In Proceedings of the Third Regional Roundtable: Refractory, Process Industry, Nanotechnologies and Nanomedicine ROSOV PINN, Belgrade, Serbia, 1–2 June 2017. [Google Scholar]
- Putteeraj, M.; Lim, W.L.; Teoh, S.L.; Yahaya, M.F. Flavonoids and its neuroprotective effects on brain ischemia and neurodegenerative diseases. Curr. Drug Targets 2018, 19, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Hosseinzadeh, H.; Nassiri-Asl, M. Neuroprotective effects of flavonoids in epilepsy. In Neuroprotective Natural Products; Wiley: Hoboken, NJ, USA, 2017; pp. 279–291. [Google Scholar]
- Szwajgier, D.; Borowiec, K.; Pustelniak, K. The neuroprotective effects of phenolic acids: Molecular mechanism of action. Nutrients 2017, 9, 477. [Google Scholar] [CrossRef] [Green Version]
- González-Sarrías, A.; Núñez-Sánchez, M.Á.; Tomás-Barberán, F.A.; Espín, J.C. Neuroprotective effects of bioavailable polyphenol-derived metabolites against oxidative stress-induced cytotoxicity in human neuroblastoma sh-sy5y cells. J. Agric. Food Chem. 2017, 65, 752–758. [Google Scholar] [CrossRef] [Green Version]
- Tair, K.; Kharoubi, O.; Tair, O.A.; Hellal, N.; Benyettou, I.; Aoues, A. Aluminium-induced acute neurotoxicity in rats: Treatment with aqueous extract of Arthrophytum (Hammada scoparia). J. Acute Dis. 2016, 5, 470–482. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, M.R.; Ferreira, G.C.; Schuck, P.F. Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in sh-sy5y cells: Role for pi3k/akt/nrf2 pathway. Toxicol. Vitr. 2016, 32, 41–54. [Google Scholar] [CrossRef]
- Lee, B.; Yeom, M.; Shim, I.; Lee, H.; Hahm, D.H. Protective Effects of Quercetin on Anxiety-Like Symptoms and Neuroinflammation Induced by Lipopolysaccharide in Rats. Evid.-Based Complement. Altern. Med. 2020, 2020, 4892415. [Google Scholar] [CrossRef]
- Selvakumar, K.; Bavithra, S.; Suganthi, M.; Benson, C.S.; Elumalai, P.; Arunkumar, R.; Krishnamoorthy, G.; Venkataraman, P.; Arunakaran, J. Protective role of quercetin on PCBs-induced oxidative stress and apoptosis in hippocampus of adult rats. Neurochem. Res. 2012, 37, 708–721. [Google Scholar] [CrossRef]
- Nkpaa, K.W.; Onyeso, G.I. Rutin attenuates neurobehavioral deficits, oxidative stress, neuro-inflammation and apoptosis in fluoride treated rats. Neurosci. Lett. 2018, 682, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ali, A.; Ali, J.; Sahni, J.; Baboota, S. Rutin: Therapeutic potential and recent advances in drug delivery. Expert Opin. Investig. Drugs 2013, 22, 1063–1079. [Google Scholar] [CrossRef]
- Spencer, J.P. Flavonoids: Modulators of brain function? Br. J. Nutr. 2008, 99, 60–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, A.Y.; Wang, Q.; Simonyi, A.; Sun, G.Y. Botanical phenolics and brain health. NeuroMolecular Med. 2008, 10, 259–274. [Google Scholar] [CrossRef] [Green Version]
- Yammine, A.; Nury, T.; Vejux, A.; Latruffe, N.; Vervandier-Fasseur, D.; Samadi, M.; Greige-Gerges, H.; Auezova, L.; Lizard, G. Prevention of 7-Ketocholesterol-Induced Overproduction of Reactive Oxygen Species, Mitochondrial Dysfunction and Cell Death with Major Nutrients (Polyphenols, ω3 and ω9 Unsaturated Fatty Acids) of the Mediterranean Diet on N2a Neuronal Cells. Molecules 2020, 25, 2296. [Google Scholar] [CrossRef] [PubMed]
- Hallal, N.; Kharoubi, O.; Benyettou, I.; Tair, K.; Ozaslan, M.; Aoues, A. In vivo Amelioration of Oxidative Stress by Artemisia absinthium L. Administration on Mercuric Chloride Toxicity in Brain Regions. J. Biol. Sci. 2016, 16, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; Scacco, S.; Sardanelli, A.M.; Vergari, R.; Papa, F.; Budde, S.; van den Heuvel, L.; Smeitink, J. Mutation in the NDUFS4 gene of complex I abolishes cAMPdependent activation of the complex in a child with fatal neurological syndrome. FEBS Lett. 2001, 489, 259–262. [Google Scholar] [CrossRef]
- Deckel, A.W. Nitric oxide and nitric oxide synthase in Huntington’s disease. J. Neurosci. Res. 2001, 64, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento-Dos-Santos, G.; de-Souza-Ferreira, E.; Linden, R.; Galina, A.; Petrs-Silva, H. Mitotherapy: Unraveling a Promising Treatment for Disorders of the Central Nervous System and Other Systemic Conditions. Cells 2021, 10, 1827. [Google Scholar] [CrossRef] [PubMed]
- Yammine, A.; Zarrouk, A.; Nury, T.; Vejux, A.; Latruffe, N.; Vervandier-Fasseur, D.; Samadi, M.; Mackrill, J.J.; Greige-Gerges, H.; Auezova, L.; et al. Prevention by Dietary Polyphenols (Resveratrol, Quercetin, Apigenin) Against 7-Ketocholesterol-Induced Oxiapoptophagy in Neuronal N2a Cells: Potential Interest for the Treatment of Neurodegenerative and Age-Related Diseases. Cells 2020, 9, 2346. [Google Scholar] [CrossRef]
- Nury, T.; Yammine, A.; Ghzaiel, I.; Sassi, K.; Zarrouk, A.; Brahmi, F.; Samadi, M.; Rup-Jacques, S.; Vervandier-Fasseur, D.; Pais de Barros, J.P.; et al. Attenuation of 7-ketocholesterol- and 7β-hydroxycholesterol-induced oxiapoptophagy by nutrients, synthetic molecules and oils: Potential for the prevention of age-related diseases. Ageing Res. Rev. 2021, 68, 101324. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kessas, K.; Chouari, Z.; Ghzaiel, I.; Zarrouk, A.; Ksila, M.; Ghrairi, T.; El Midaoui, A.; Lizard, G.; Kharoubi, O. Role of Bioactive Compounds in the Regulation of Mitochondrial Dysfunctions in Brain and Age-Related Neurodegenerative Diseases. Cells 2022, 11, 257. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020257
Kessas K, Chouari Z, Ghzaiel I, Zarrouk A, Ksila M, Ghrairi T, El Midaoui A, Lizard G, Kharoubi O. Role of Bioactive Compounds in the Regulation of Mitochondrial Dysfunctions in Brain and Age-Related Neurodegenerative Diseases. Cells. 2022; 11(2):257. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020257
Chicago/Turabian StyleKessas, Khadidja, Zhor Chouari, Imen Ghzaiel, Amira Zarrouk, Mohamed Ksila, Taoufik Ghrairi, Adil El Midaoui, Gérard Lizard, and Omar Kharoubi. 2022. "Role of Bioactive Compounds in the Regulation of Mitochondrial Dysfunctions in Brain and Age-Related Neurodegenerative Diseases" Cells 11, no. 2: 257. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020257