
RNA Molecular Signature Profiling in PBMCs of Sporadic ALS Patients: HSP70 Overexpression Is Associated with Nuclear SOD1

 , , ,
, , ,  , , , ,
, , , ,  , , and
, , and
Abstract
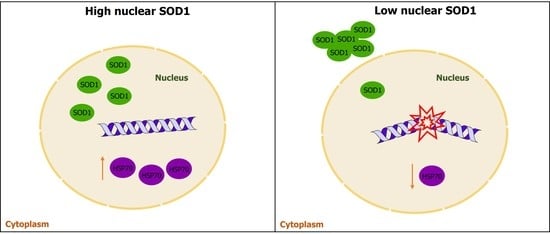
:
1. Introduction
2. Materials and Methods
2.1. Patient Enrollment
2.2. PBMC Isolation from Blood Samples
2.3. Subcellular Fractionation
2.4. Western Blotting Analysis
2.5. Selection of a Cut-Off Value
2.6. RNA Extraction
2.7. Library Preparation for RNA-seq and Bioinformatic Data Analysis
2.8. Pathway Analysis
2.9. Real-Time PCR
2.10. Comet Assay
2.11. Immunofluorescence
3. Results
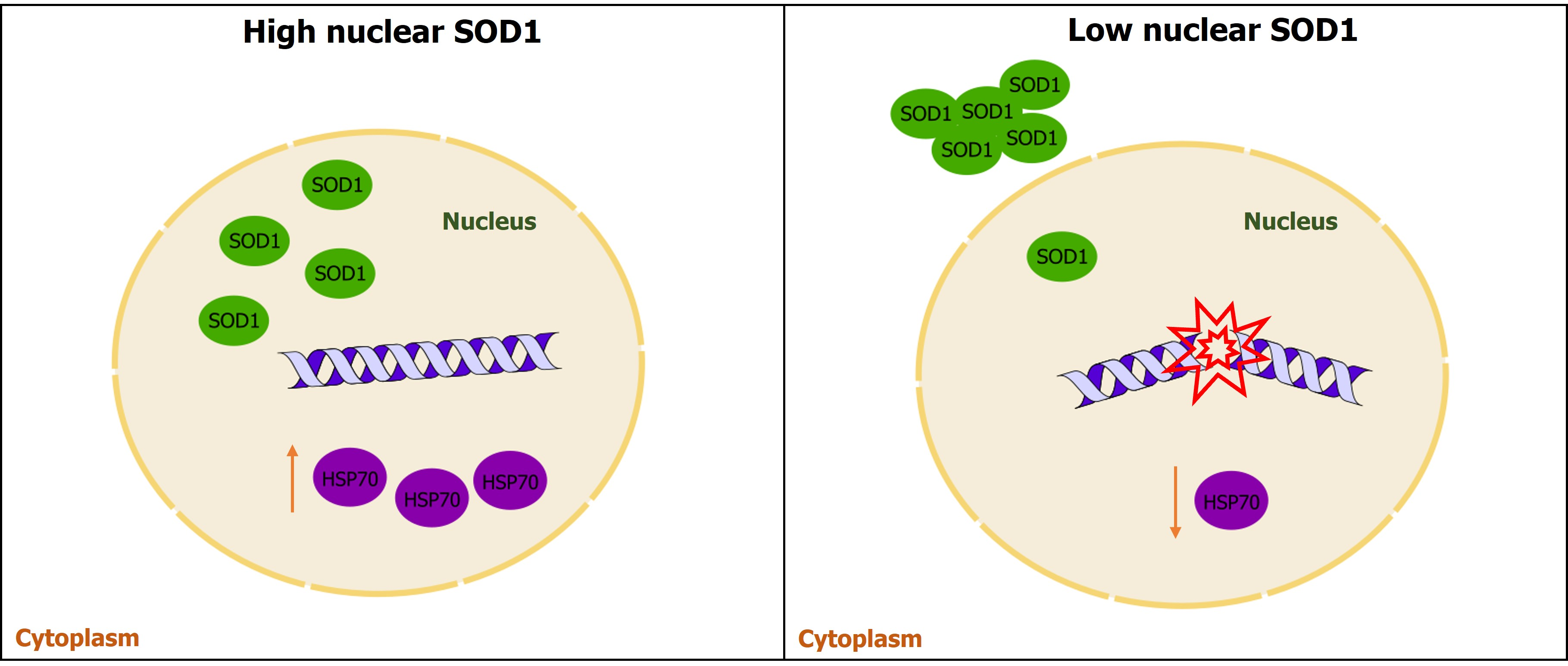
3.1. Whole Transcriptome Analysis in PBMCs of High- and Low-nSOD6 sALS Patients and Healthy Matched Controls
3.1.1. Classification of sALS Patients with High and Low Nuclear SOD1
3.1.2. Whole Transcriptome Analysis in PBMCs of High- and Low-nSOD1 sALS Patients and Healthy Controls
3.2. Evaluation of Histone 3 Methylation
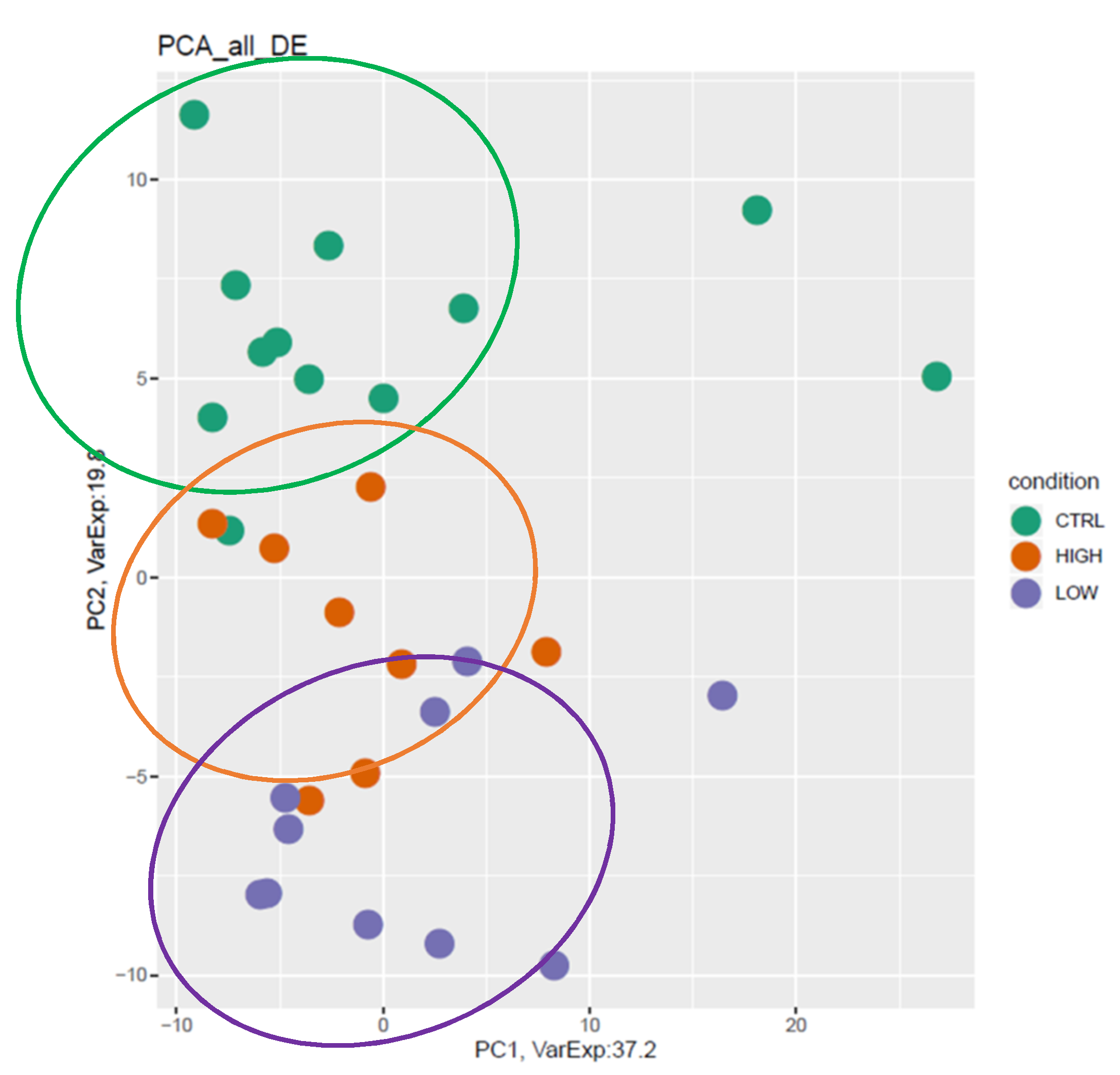
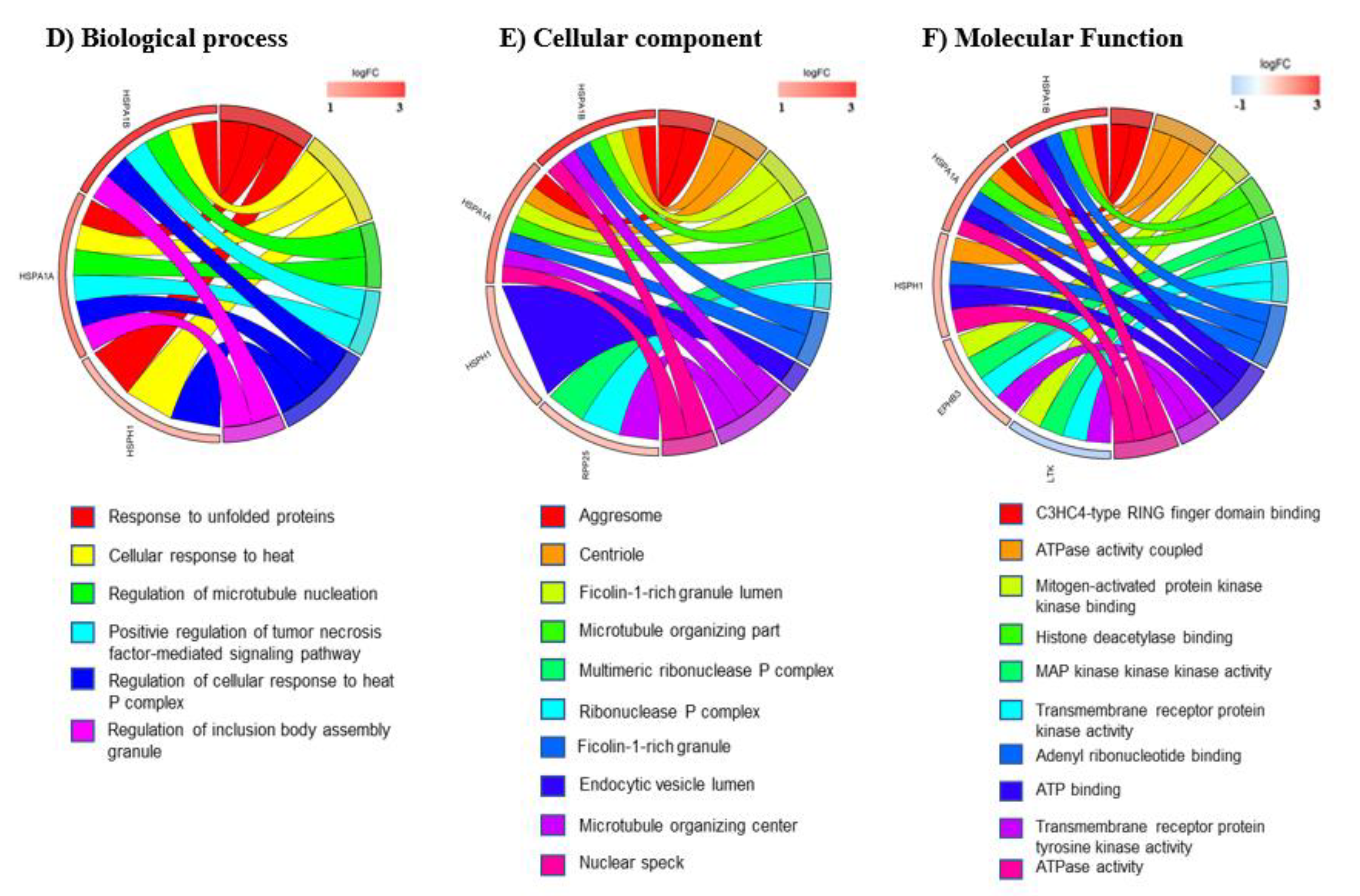
3.3. mRNA Pathway Analysis
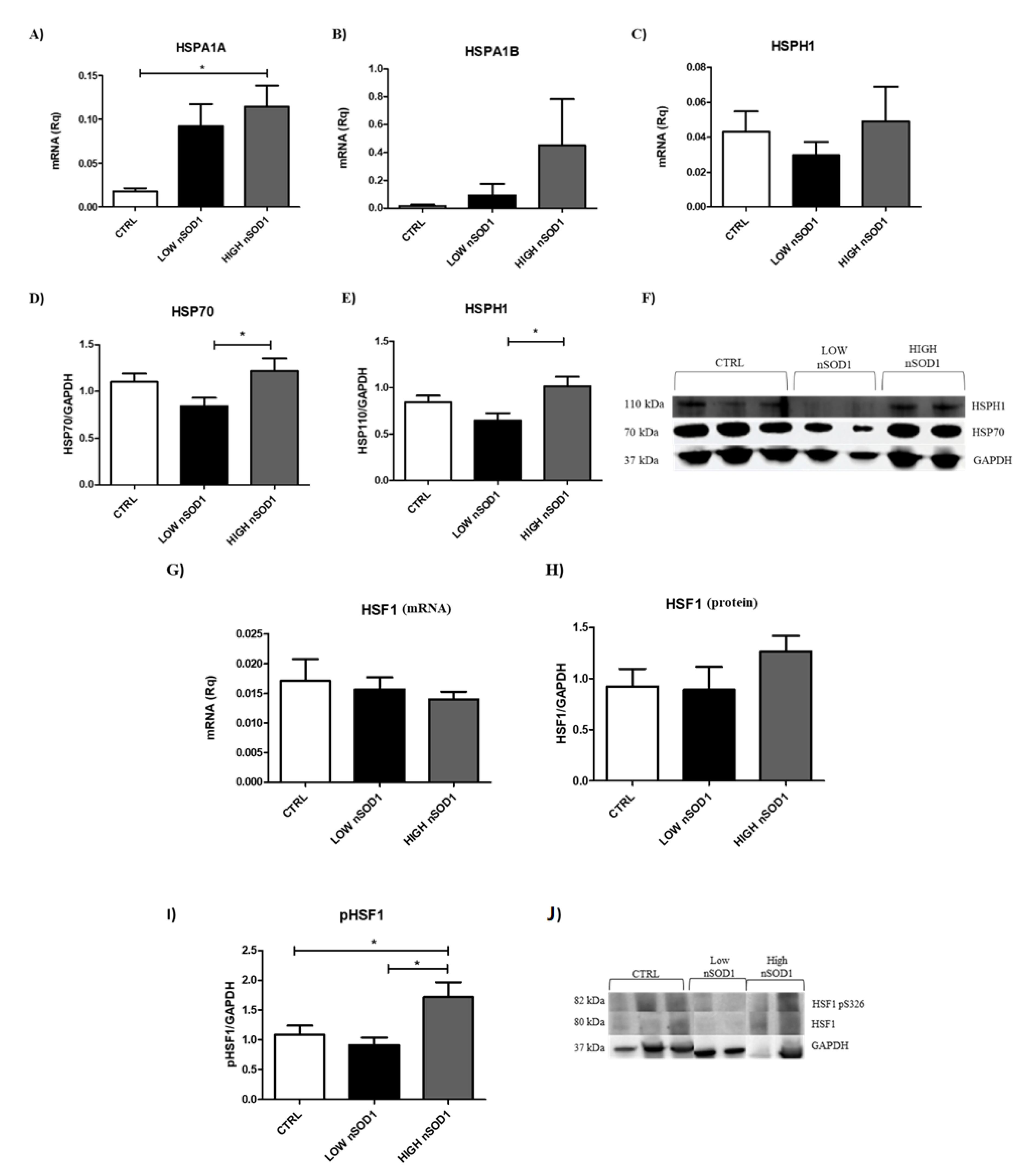
3.4. Heat Shock Proteins and DNA Damage Evaluation in PBMCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide Dismutase Multigene Family: A Comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordoni, M.; Pansarasa, O.; Dell’Orco, M.; Crippa, V.; Gagliardi, S.; Sproviero, D.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Tedeschi, G.; et al. Nuclear Phospho-SOD1 Protects DNA from Oxidative Stress Damage in Amyotrophic Lateral Sclerosis. J. Clin. Med. 2019, 8, 729. [Google Scholar] [CrossRef] [Green Version]
- Pansarasa, O.; Bordoni, M.; Drufuca, L.; Diamanti, L.; Sproviero, D.; Trotti, R.; Bernuzzi, S.; Salvia, S. La; Gagliardi, S.; Ceroni, M.; et al. Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. DMM Dis. Model. Mech. 2018, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Wang, J.; Henderson, M.J.; Yang, P.; Hagen, B.M.; Siddique, T.; Vogel, B.E.; Deng, H.-X.; Fang, S. Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. eLife 2017, 6, e23759. [Google Scholar] [CrossRef] [PubMed]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. Sod1 in amyotrophic lateral sclerosis: “ambivalent” behavior connected to the disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [Green Version]
- Cova, E.; Cereda, C.; Galli, A.; Curti, D.; Finotti, C.; Di Poto, C.; Corato, M.; Mazzini, G.; Ceroni, M. Modified expression of Bcl-2 and SOD1 proteins in lymphocytes from sporadic ALS patients. Neurosci. Lett. 2006, 399, 186–190. [Google Scholar] [CrossRef]
- Gagliardi, S.; Cova, E.; Davin, A.; Guareschi, S.; Abel, K.; Alvisi, E.; Laforenza, U.; Ghidoni, R.; Cashman, J.R.; Ceroni, M.; et al. SOD1 mRNA expression in sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2010, 39, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K. wa.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [Green Version]
- Guareschi, S.; Cova, E.; Cereda, C.; Ceroni, M.; Donetti, E.; Bosco, D.A.; Trotti, D.; Pasinelli, P. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 2012, 109, 5074–5079. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, Y.; Yang, H.; Xu, Y.; Zhou, X.; Zhang, X.; Xie, Z.; Bi, J. The effect of BACE1-AS on β-amyloid generation by regulating BACE1 mRNA expression. BMC Mol. Biol. 2019, 20, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered Intracellular Localization of SOD1 in Leukocytes from Patients with Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e75916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasil, A.d.A.; de Carvalho, M.D.C.; Gerhardt, E.; Queiroz, D.D.; Pereira, M.D.; Outeiro, T.F.; Eleutherio, E.C.A. Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1. Proc. Natl. Acad. Sci. USA 2019, 116, 25991–26000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, E.; Matthias, P.; Miiller, M.M.; Schaffner, W. Rapid Detection of Octamer Binding Proteins with “Mini-Extracts”, Prepared from a Small Number of Cells. Nucleic Acids Res. 1989, 17, 6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Z.; Calin, G.A.; de Paula, H.M.; Medeiros, L.J.; Fernandez, M.H.; Shimizu, M.; Garcia-Manero, G.; Bueso-Ramos, C.E. Circulating microRNAs let-7a and miR-16 predict progression-free survival and overall survival in patients with myelodysplastic syndrome. Blood 2011, 118, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.G.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Końca, K.; Lankoff, A.; Banasik, A.; Lisowska, H.; Kuszewski, T.; Góźdź, S.; Koza, Z.; Wojcik, A. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res.-Genet. Toxicol. Environ. Mutagen. 2003, 534, 15–20. [Google Scholar] [CrossRef]
- Zucca, S.; Gagliardi, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. RNA-seq profiling in peripheral blood mononuclear cells of amyotrophic lateral sclerosis patients and controls. Sci. Data 2019, 6, 190006. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, Y.; Loh, Y.P.; Tng, J.Q.; Lim, M.C.; Cao, Z.; Raju, A.; Lieberman Aiden, E.; Li, S.; Manikandan, L.; et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat. Commun. 2021, 12, 719. [Google Scholar] [CrossRef] [PubMed]
- Igolkina, A.A.; Zinkevich, A.; Karandasheva, K.O.; Popov, A.A.; Selifanova, M.V.; Nikolaeva, D.; Tkachev, V.; Penzar, D.; Nikitin, D.M.; Buzdin, A. H3K4me3, H3K9ac, H3K27ac, H3K27me3 and H3K9me3 Histone Tags Suggest Distinct Regulatory Evolution of Open and Condensed Chromatin Landmarks. Cells 2019, 8, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrez, L.; Causse, S.; Borges Bonan, N.; Dumétier, B.; Garrido, C. Heat-shock proteins: Chaperoning DNA repair. Oncogene 2020, 39, 516–529. [Google Scholar] [CrossRef]
- Barna, J.; Csermely, P.; Vellai, T. Roles of heat shock factor 1 beyond the heat shock response. Cell. Mol. Life Sci. 2018, 75, 2897–2916. [Google Scholar] [CrossRef]
- Vousden, K.H. Outcomes of p53 activation—Spoilt for choice. J. Cell Sci. 2006, 119, 5015–5020. [Google Scholar] [CrossRef] [Green Version]
- Kupershmit, I.; Khoury-Haddad, H.; Awwad, S.W.; Guttmann-Raviv, N.; Ayoub, N. KDM4C (GASC1) lysine demethylase is associated with mitotic chromatin and regulates chromosome segregation during mitosis. Nucleic Acids Res. 2014, 42, 6168–6182. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wang, B.; Lee, W.Y.-W.; Pong, U.K.; Leung, K.T.; Li, X.; Liu, Z.; Chen, R.; Lin, J.C.; Tsang, L.L.; et al. KDM3A and KDM4C Regulate Mesenchymal Stromal Cell Senescence and Bone Aging via Condensin-mediated Heterochromatin Reorganization. iScience 2019, 21, 375–390. [Google Scholar] [CrossRef]
- Mayer, M.P. Gymnastics of molecular chaperones. Mol. Cell 2010, 39, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Huang, S.; Yang, J.; Niu, P.; Gong, Z.; Liu, X.; Xin, L.; Currie, R.W.; Wu, T. HspA1A facilitates DNA repair in human bronchial epithelial cells exposed to Benzo[a]pyrene and interacts with casein kinase 2. Cell Stress Chaperones 2014, 19, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, C.R.; Dix, D.J.; Sharma, G.G.; Pandita, R.K.; Gupta, A.; Funk, M.; Pandita, T.K. Genomic Instability and Enhanced Radiosensitivity in Hsp70.1- and Hsp70.3-Deficient Mice. Mol. Cell. Biol. 2004, 24, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandle, A.T.; Mazzanti, C.; Alexander, H.R.; Roberts, D.D.; Libutti, S.K. Endothelial monocyte activating polypeptide-II induced gene expression changes in endothelial cells. Cytokine 2005, 30, 347–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Hallstrom, T.C. Sensitivity to TOP2 targeting chemotherapeutics is regulated by Oct1 and FILIP1L. PLoS ONE 2012, 7, e42921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguila, B.; Morris, A.B.; Spina, R.; Bar, E.; Schraner, J.; Vinkler, R.; Sohn, J.W.; Welford, S.M. The Ig superfamily protein PTGFRN coordinates survival signaling in glioblastoma multiforme. Cancer Lett. 2019, 462, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Serlidaki, D.; van Waarde, M.A.W.H.; Rohland, L.; Wentink, A.S.; Dekker, S.L.; Kamphuis, M.J.; Boertien, J.M.; Brunsting, J.F.; Nillegoda, N.B.; Bukau, B.; et al. Functional diversity between HSP70 paralogs caused by variable interactions with specific co-chaperones. J. Biol. Chem. 2020, 295, 7301–7316. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nat. Commun. 2014, 5, 5513. [Google Scholar] [CrossRef] [Green Version]
- Katsogiannou, M.; Andrieu, C.; Baylot, V.; Baudot, A.; Dusetti, N.J.; Gayet, O.; Finetti, P.; Garrido, C.; Birnbaum, D.; Bertucci, F.; et al. The functional landscape of Hsp27 reveals new cellular processes such as DNA repair and alternative splicing and proposes novel anticancer targets. Mol. Cell. Proteom. 2014, 13, 3585–3601. [Google Scholar] [CrossRef] [Green Version]
- Sottile, M.L.; Nadin, S.B. Heat shock proteins and DNA repair mechanisms: An updated overview. Cell Stress Chaperones 2018, 23, 303–315. [Google Scholar] [CrossRef]
- Sorci, G.; Bianchi, R.; Riuzzi, F.; Tubaro, C.; Arcuri, C.; Giambanco, I.; Donato, R. S100B protein, a damage-associated molecular pattern protein in the brain and heart, and beyond. Cardiovasc. Psychiatry Neurol. 2010, 2010, 656481. [Google Scholar] [CrossRef] [PubMed]
- Juranek, J.K.; Daffu, G.K.; Wojtkiewicz, J.; Lacomis, D.; Kofler, J.; Schmidt, A.M. Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2015, 9, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.-H.; Kim, H.-M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Xu, W.; Lan, F. Histone lysine demethylases in mammalian embryonic development. Exp. Mol. Med. 2017, 49, e325. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age | Sex | Onset | SOD1/PCNA |
| M53 | 53 | M | Spinal | 1.935 |
| F69 | 69 | F | Spinal | 1.673 |
| M69 | 69 | M | Spinal | 2.021 |
| F69 | 69 | F | Spinal | 1.807 |
| F64 | 64 | F | Spinal | 2.903 |
| M58 | 58 | M | Spinal | 4.456 |
| M44 | 44 | M | Spinal | 3.395 |
| F58 | 58 | F | Bulbar | 1.566 |
| F66 | 66 | F | Spinal | 1.195 |
| M65 | 65 | M | Bulbar | 1.319 |
| M68 | 68 | M | Spinal | 1.275 |
| F71 | 71 | F | Spinal | 0.718 |
| F74 | 74 | F | Bulbar | 0.319 |
| F70 | 70 | F | Spinal | 0.539 |
| M66 | 66 | M | Spinal | 1.149 |
| M72 | 72 | M | Spinal | 0.624 |
| F89 | 89 | F | Bulbar | 0.492 |
| F76 | 76 | F | Bulbar | 0.222 |
| Control | Age | Sex | Onset | SOD1/PCNA |
| M68 | 68 | M | / | 2.334 |
| M64 | 64 | M | / | 1.300 |
| M63 | 63 | M | / | 0.909 |
| M61 | 61 | M | / | 4.203 |
| M60 | 60 | M | / | 2.088 |
| F58 | 58 | F | / | 1.031 |
| M57 | 57 | M | / | 0.404 |
| M56 | 56 | M | / | 1.692 |
| F51 | 51 | F | / | 0.151 |
| M51 | 51 | M | / | 0.635 |
| M46 | 46 | M | / | 3.075 |
| M45 | 45 | M | / | 2.955 |
| Low nSOD1 | High nSOD1 | |||||
|---|---|---|---|---|---|---|
| mRNA | lncRNA | Other | mRNA | lncRNA | Other | |
| UP | 20 | 9 | 11 | 12 | 2 | 3 |
| DOWN | 15 | 5 | 2 | 3 | 2 | 3 |
| Subtotal | 35 | 14 | 13 | 15 | 4 | 6 |
| Total | 62 | 25 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garofalo, M.; Pandini, C.; Bordoni, M.; Jacchetti, E.; Diamanti, L.; Carelli, S.; Raimondi, M.T.; Sproviero, D.; Crippa, V.; Carra, S.; et al. RNA Molecular Signature Profiling in PBMCs of Sporadic ALS Patients: HSP70 Overexpression Is Associated with Nuclear SOD1. Cells 2022, 11, 293. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020293
Garofalo M, Pandini C, Bordoni M, Jacchetti E, Diamanti L, Carelli S, Raimondi MT, Sproviero D, Crippa V, Carra S, et al. RNA Molecular Signature Profiling in PBMCs of Sporadic ALS Patients: HSP70 Overexpression Is Associated with Nuclear SOD1. Cells. 2022; 11(2):293. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020293
Chicago/Turabian StyleGarofalo, Maria, Cecilia Pandini, Matteo Bordoni, Emanuela Jacchetti, Luca Diamanti, Stephana Carelli, Manuela Teresa Raimondi, Daisy Sproviero, Valeria Crippa, Serena Carra, and et al. 2022. "RNA Molecular Signature Profiling in PBMCs of Sporadic ALS Patients: HSP70 Overexpression Is Associated with Nuclear SOD1" Cells 11, no. 2: 293. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020293