
Targeted Inhibition of Matrix Metalloproteinase-8 Prevents Aortic Dissection in a Murine Model
 , ,
, ,
Abstract
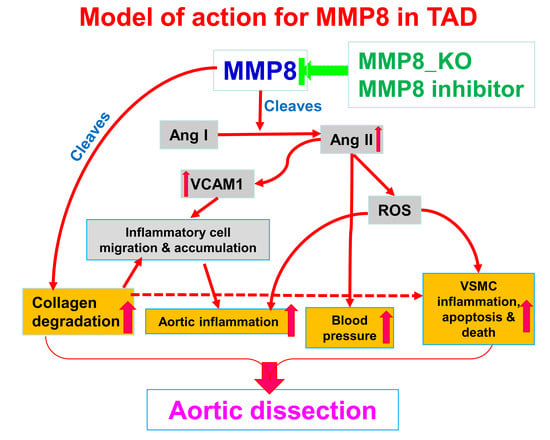
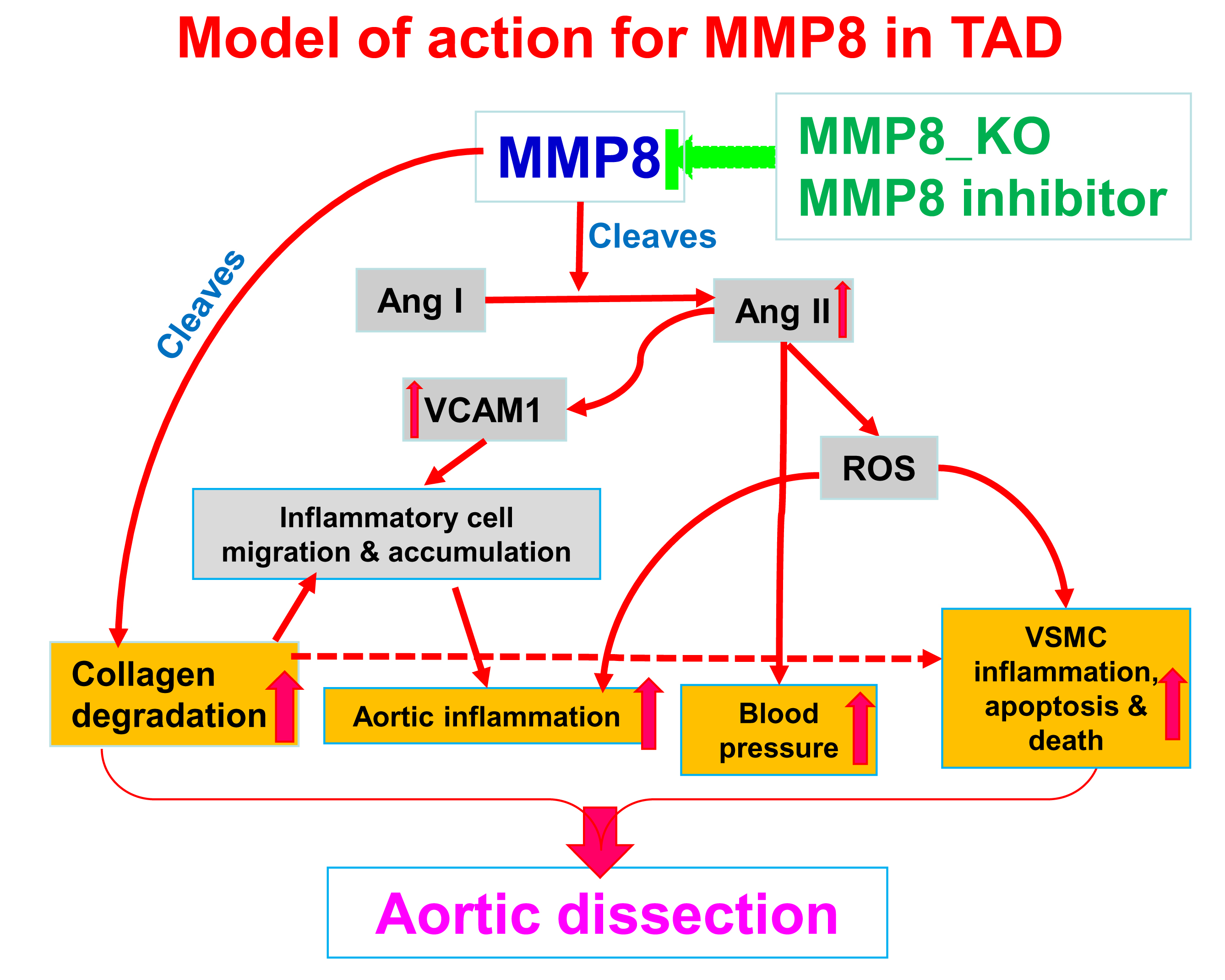
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Animal Experiments, Anesthesia, and Euthanasia
2.3. Collection of Human Aorta Tissue Specimens and Sera
2.4. Histopathological Analysis
2.5. Blood Pressure Measurement
2.6. Aortic Tissue Immunofluorescence Staining
2.7. TUNEL Staining for SMC Apoptosis
2.8. Bone Marrow Cell Isolation
2.9. C166 Culture and MISSION esiRNA Transfection
2.10. Murine Aortic SMC Culture and Treatments
2.11. Real-Time Quantitative PCR (RT-qPCR)
2.12. ELISA Analysis
2.13. MMP8 Activity Analysis
2.14. ROS Measurement
2.15. Cell Invasion and Transendothelial Migration Assay
2.16. Cell Viability Analyses
2.17. Statistical Analysis
3. Results
3.1. MMP8 Expression Was Increased during BAPN-Induced TAD Development
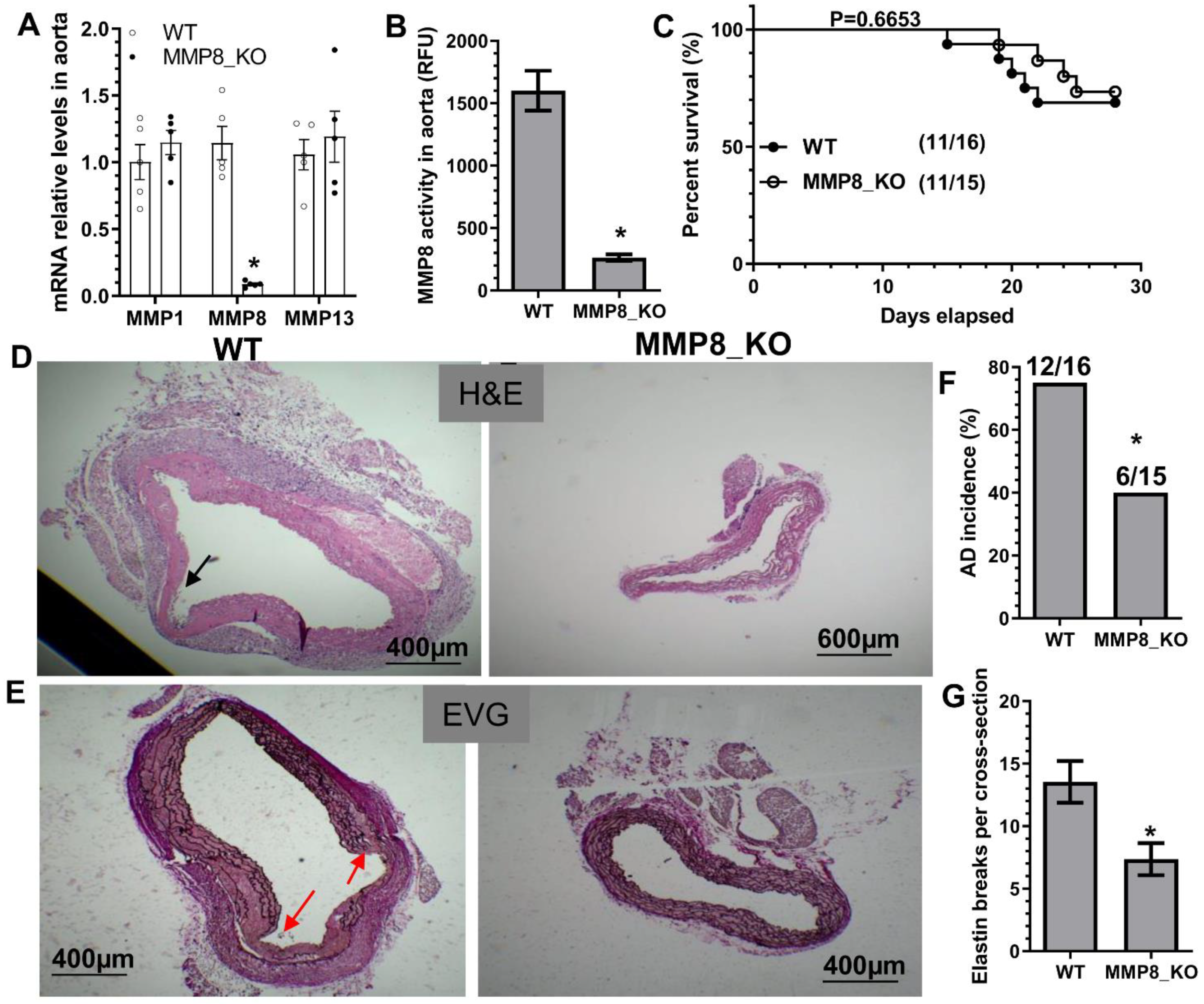
3.2. MMP8 Deficiency Inhibited BAPN-Induced TAD
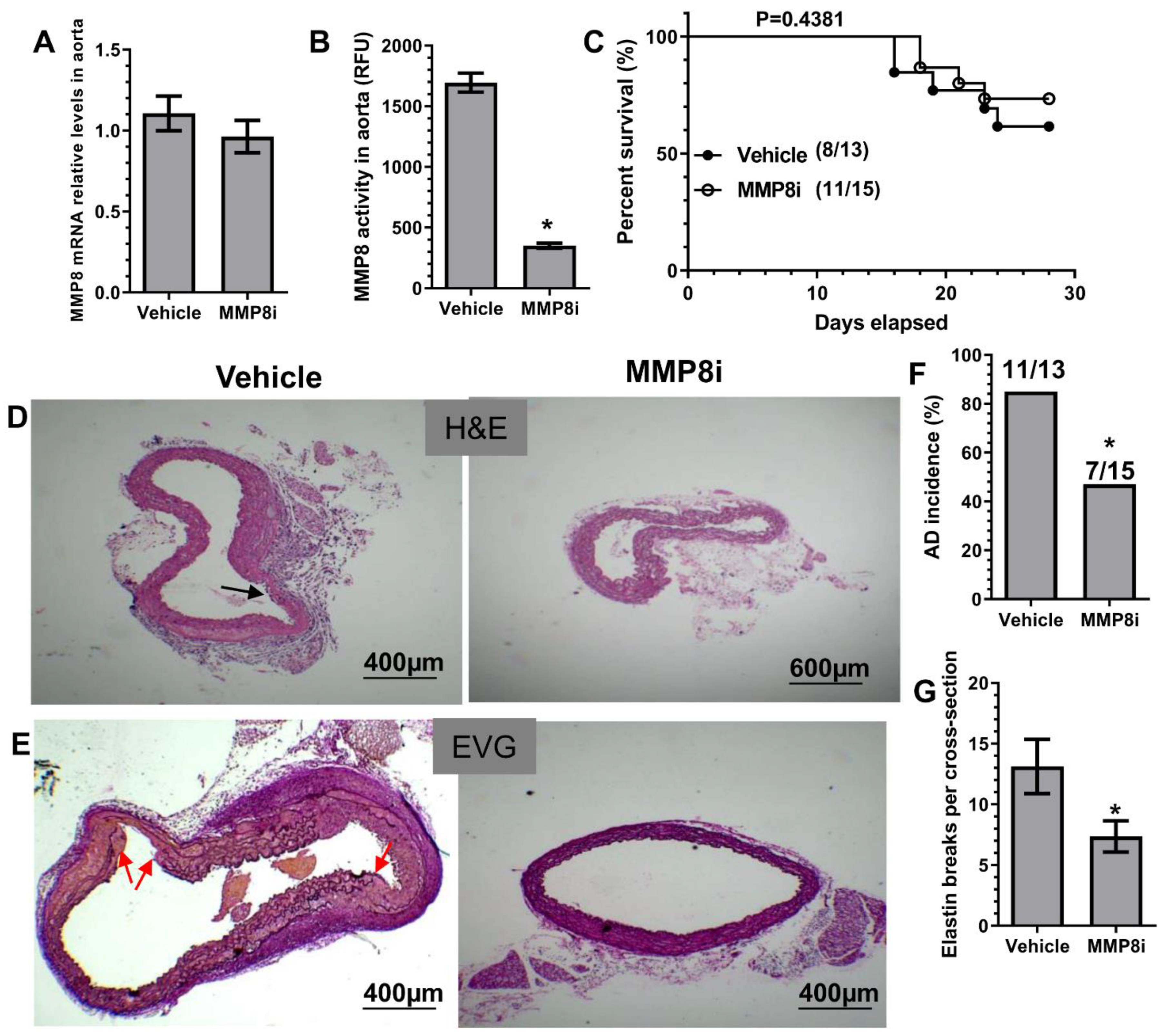
3.3. Pharmacological Inhibition of MMP8 Decreased TAD
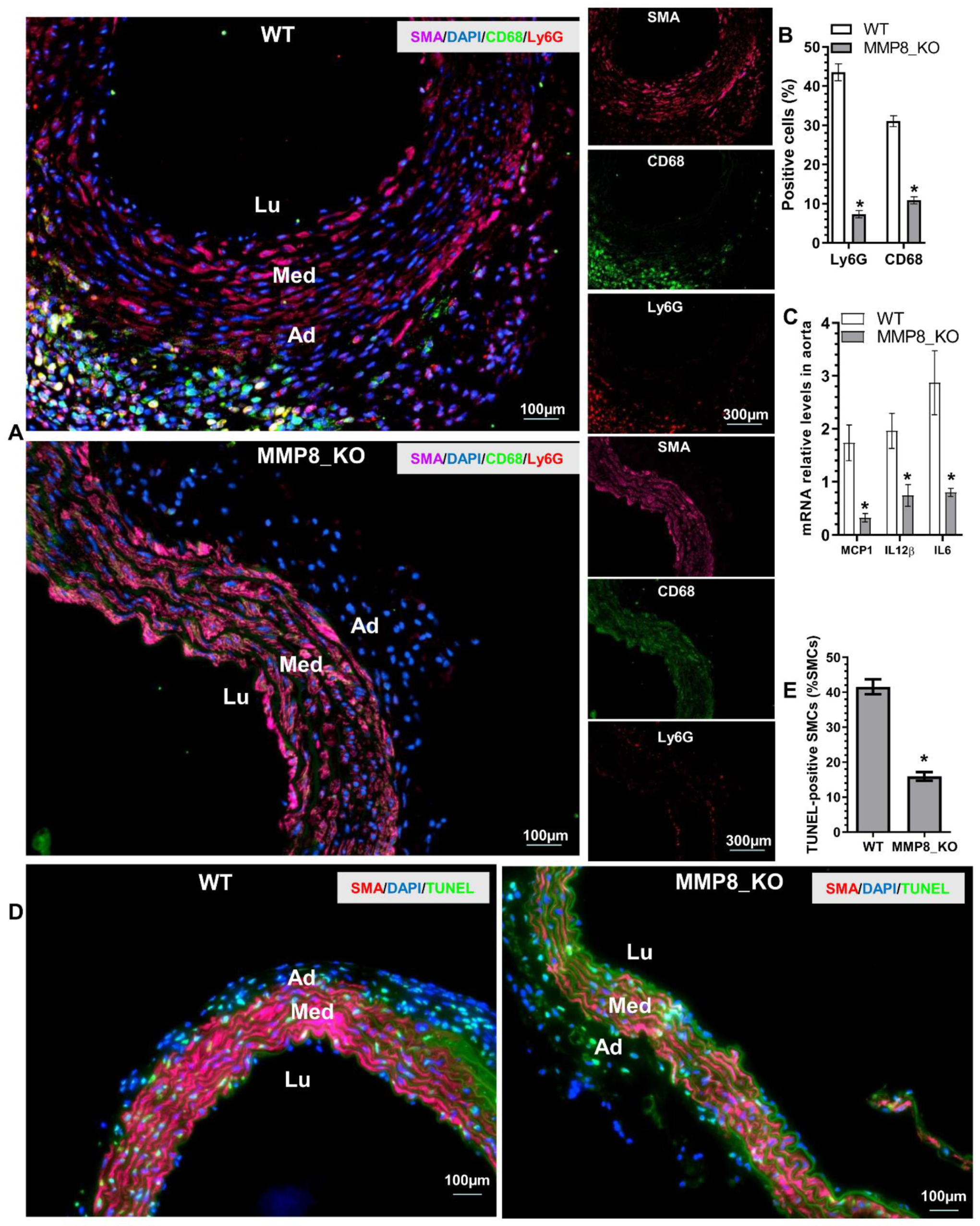
3.4. Aortic Inflammatory Cell Accumulation and SMC Apoptosis Was Reduced in MMP8_KO Mice
3.5. Reduced Ang II Levels, Lower Blood Pressure, and Decreased VCAM-1 Expressions Were Observed in MMP8_KO Mice
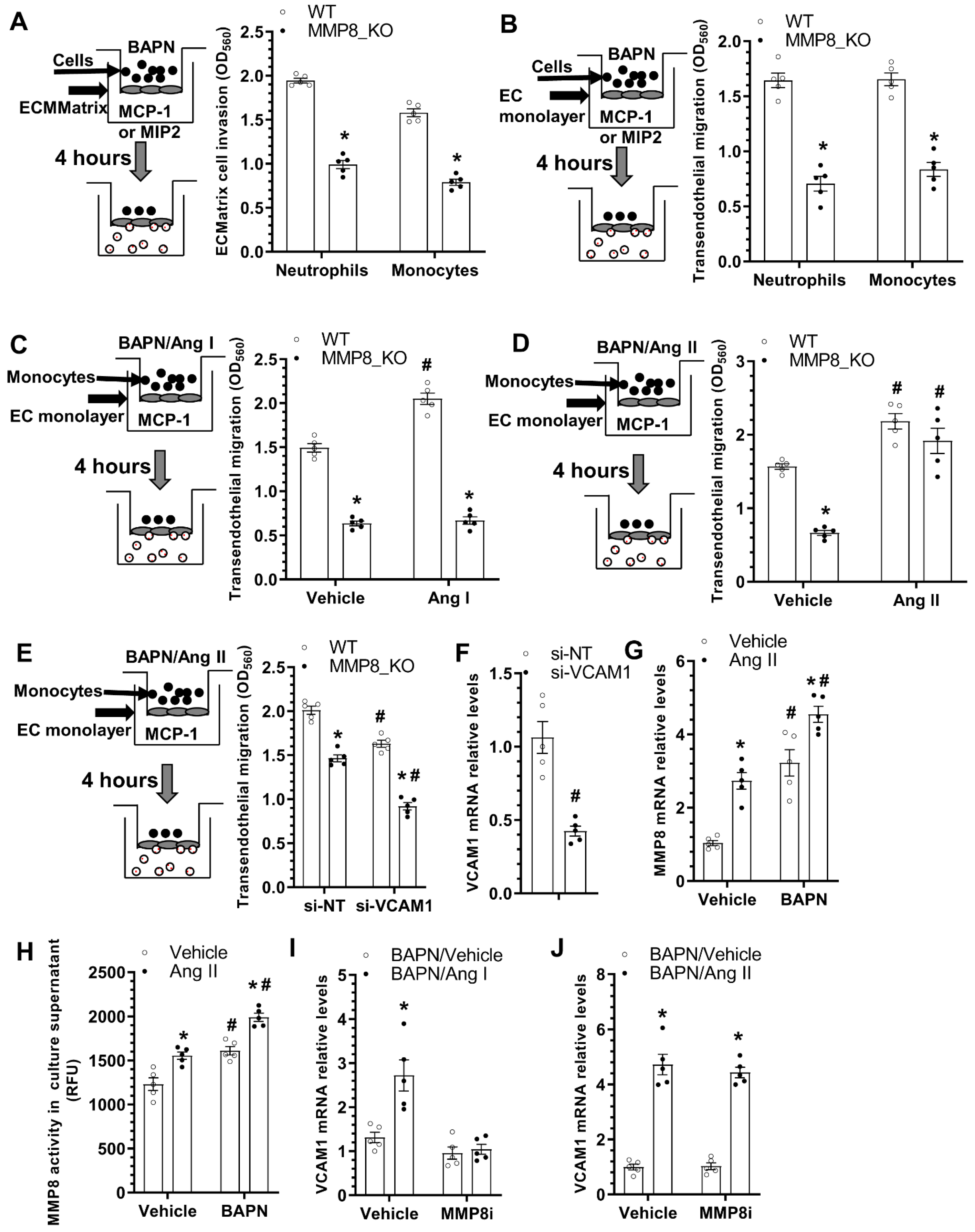
3.6. MMP8 Augmented Inflammatory Cell Invasion and Transendothelial Migration
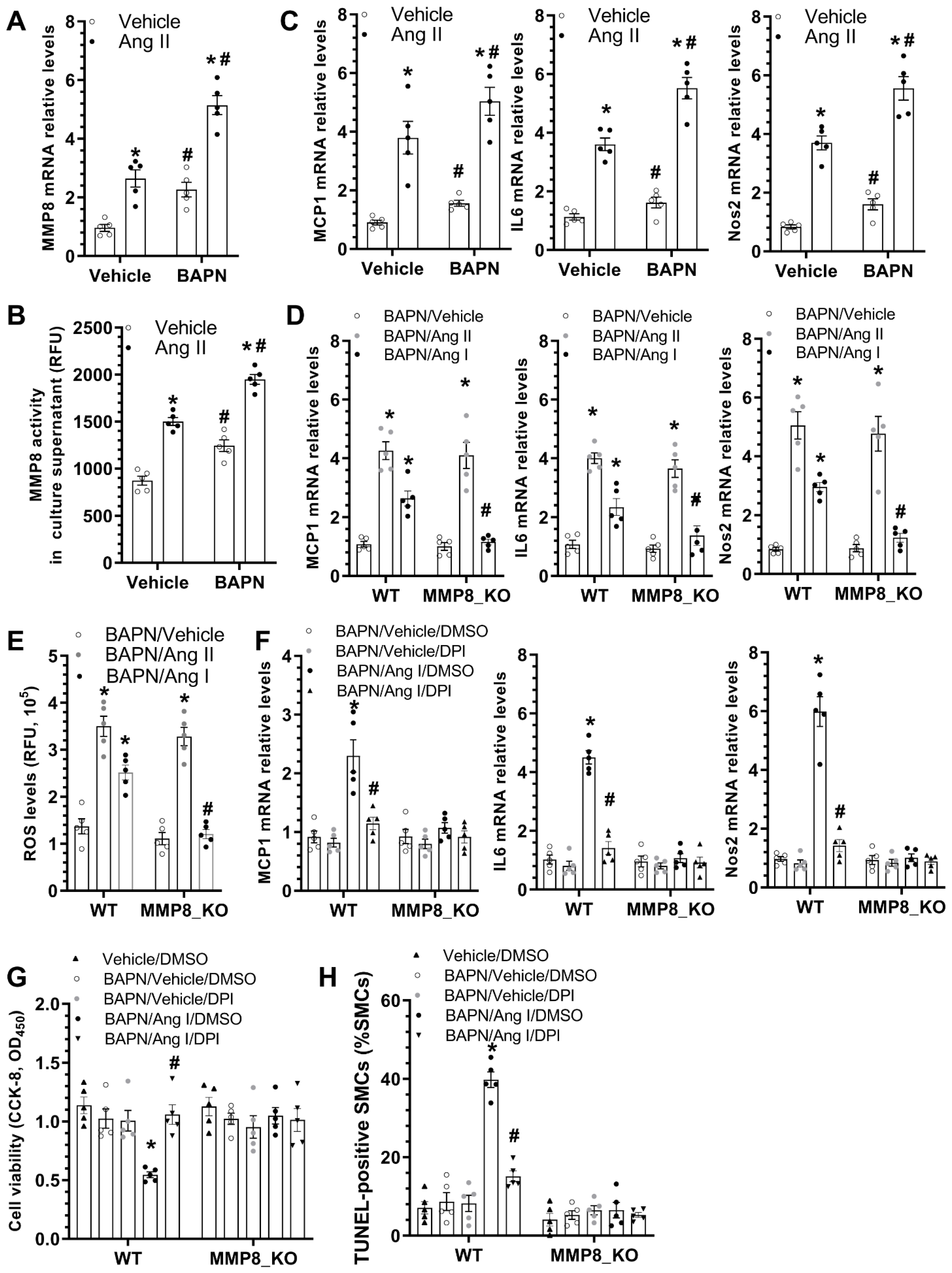
3.7. MMP8 Mediated BAPN/Ang II-Induced SMC Inflammation and Apoptosis
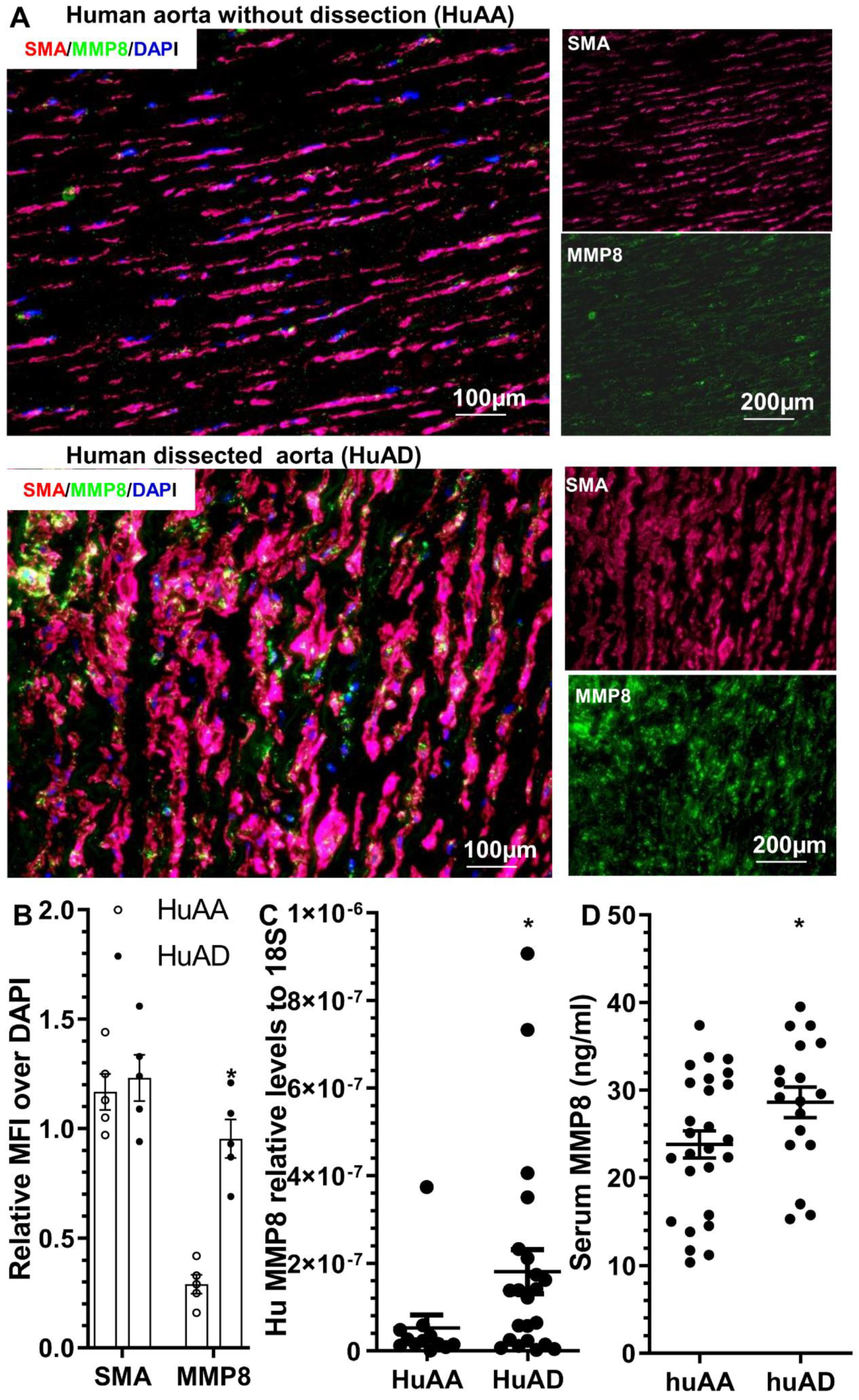
3.8. MMP8 Is Increased in the Dissected Human Arteries and Serum from Patients with Acute TAD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mussa, F.F.; Horton, J.D.; Moridzadeh, R.; Nicholson, J.; Trimarchi, S.; Eagle, K.A. Acute Aortic Dissection and Intramural Hematoma: A Systematic Review. JAMA 2016, 316, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Nienaber, C.A.; Clough, R.E. Management of acute aortic dissection. Lancet 2015, 385, 800–811. [Google Scholar] [CrossRef]
- Hagan, P.G.; Nienaber, C.A.; Isselbacher, E.M.; Bruckman, D.; Karavite, D.J.; Russman, P.L.; Evangelista, A.; Fattori, R.; Suzuki, T.; Oh, J.K.; et al. The International Registry of Acute Aortic Dissection (IRAD): New insights into an old disease. JAMA 2000, 283, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Shimoda, M.; Endo, J.; Kohno, T.; Katsumata, Y.; Matsuhashi, T.; Yamamoto, T.; Ito, K.; Yan, X.; Shirakawa, K.; et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ. Res. 2015, 116, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, R.; Oo, A.Y.; Xiao, Q. Matrix Metalloproteinase in Abdominal Aortic Aneurysm and Aortic Dissection. Pharmaceuticals 2019, 12, 118. [Google Scholar] [CrossRef] [Green Version]
- Golledge, J.; Eagle, K.A. Acute aortic dissection. Lancet 2008, 372, 55–66. [Google Scholar] [CrossRef]
- Van Lint, P.; Libert, C. Matrix metalloproteinase-8: Cleavage can be decisive. Cytokine Growth Factor Rev. 2006, 17, 217–223. [Google Scholar] [CrossRef]
- Wen, G.; Zhang, C.; Chen, Q.; Luong le, A.; Mustafa, A.; Ye, S.; Xiao, Q. A Novel Role of Matrix Metalloproteinase-8 in Macrophage Differentiation and Polarization. J. Biol. Chem. 2015, 290, 19158–19172. [Google Scholar] [CrossRef] [Green Version]
- Van Den Steen, P.E.; Wuyts, A.; Husson, S.J.; Proost, P.; Van Damme, J.; Opdenakker, G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. Eur. J. Biochem. 2003, 270, 3739–3749. [Google Scholar]
- Cox, J.H.; Dean, R.A.; Roberts, C.R.; Overall, C.M. Matrix metalloproteinase processing of CXCL11/I-TAC results in loss of chemoattractant activity and altered glycosaminoglycan binding. J. Biol. Chem. 2008, 283, 19389–19399. [Google Scholar] [CrossRef] [Green Version]
- Laxton, R.C.; Hu, Y.; Duchene, J.; Zhang, F.; Zhang, Z.; Leung, K.Y.; Xiao, Q.; Scotland, R.S.; Hodgkinson, C.P.; Smith, K.; et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ. Res. 2009, 105, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhang, F.; Grassia, G.; Hu, Y.; Zhang, Z.; Xing, Q.; Yin, X.; Maddaluno, M.; Drung, B.; Schmidt, B.; et al. Matrix metalloproteinase-8 promotes vascular smooth muscle cell proliferation and neointima formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Zhang, F.; Lin, L.; Fang, C.; Wen, G.; Tsai, T.N.; Pu, X.; Sims, D.; Zhang, Z.; Yin, X.; et al. Functional role of matrix metalloproteinase-8 in stem/progenitor cell migration and their recruitment into atherosclerotic lesions. Circ. Res. 2013, 112, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S. Putative targeting of matrix metalloproteinase-8 in atherosclerosis. Pharmacol. Ther. 2015, 147, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Turu, M.M.; Krupinski, J.; Catena, E.; Rosell, A.; Montaner, J.; Rubio, F.; Alvarez-Sabin, J.; Cairols, M.; Badimon, L. Intraplaque MMP-8 levels are increased in asymptomatic patients with carotid plaque progression on ultrasound. Atherosclerosis 2006, 187, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Tuomainen, A.M.; Nyyssonen, K.; Laukkanen, J.A.; Tervahartiala, T.; Tuomainen, T.P.; Salonen, J.T.; Sorsa, T.; Pussinen, P.J. Serum matrix metalloproteinase-8 concentrations are associated with cardiovascular outcome in men. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.P.; Sukhova, G.K.; Libby, P.; Gerdes, N.; Tang, N.; Horton, D.B.; Kilbride, M.; Breitbart, R.E.; Chun, M.; Schonbeck, U. Expression of neutrophil collagenase (matrix metalloproteinase-8) in human atheroma: A novel collagenolytic pathway suggested by transcriptional profiling. Circulation 2001, 104, 1899–1904. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Wen, G.; Zhang, L.; Lin, L.; Moore, A.; Wu, S.; Ye, S.; Xiao, Q. An important role of matrix metalloproteinase-8 in angiogenesis in vitro and in vivo. Cardiovasc. Res. 2013, 99, 146–155. [Google Scholar] [CrossRef]
- Yang, F.; Chen, Q.; Yang, M.; Maguire, E.M.; Yu, X.; He, S.; Xiao, R.; Wang, C.S.; An, W.; Wu, W.; et al. Macrophage-derived MMP-8 determines smooth muscle cell differentiation from adventitia stem/progenitor cells and promotes neointima hyperplasia. Cardiovasc. Res. 2020, 116, 211–225. [Google Scholar] [CrossRef]
- Yang, K.; Ren, J.; Li, X.; Wang, Z.; Xue, L.; Cui, S.; Sang, W.; Xu, T.; Zhang, J.; Yu, J.; et al. Prevention of aortic dissection and aneurysm via an ALDH2-mediated switch in vascular smooth muscle cell phenotype. Eur. Heart J. 2020, 41, 2442–2453. [Google Scholar] [CrossRef]
- Jia, L.X.; Zhang, W.M.; Zhang, H.J.; Li, T.T.; Wang, Y.L.; Qin, Y.W.; Gu, H.; Du, J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 2015, 236, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.X.; Zhang, W.M.; Li, T.T.; Liu, Y.; Piao, C.M.; Ma, Y.C.; Lu, Y.; Wang, Y.; Liu, T.T.; Qi, Y.F.; et al. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin. Sci. 2017, 131, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Y.; Li, L.Y.; Jiao, X.L.; Jia, L.X.; Zhang, X.P.; Wang, Y.L.; Yang, S.; Li, J.; Du, J.; Wei, Y.X.; et al. Intermittent Hypoxia Alleviates beta-Aminopropionitrile Monofumarate Induced Thoracic Aortic Dissection in C57BL/6 Mice. Eur. J. Vasc. Endovasc. Surg. 2020, 59, 1000–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, I.; Cavalera, M.; Huang, S.; Su, Y.; Hanna, A.; Chen, B.; Shinde, A.V.; Conway, S.J.; Graff, J.; Frangogiannis, N.G. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 124, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Tajerian, M.; Clark, J.D. Spinal matrix metalloproteinase 8 regulates pain after peripheral trauma. J. Pain Res. 2019, 12, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.E.; Lee, E.J.; Moon, E.; Ryu, J.H.; Choi, J.W.; Kim, H.S. Matrix Metalloproteinase-8 is a Novel Pathogenetic Factor in Focal Cerebral Ischemia. Mol. Neurobiol. 2016, 53, 231–239. [Google Scholar] [CrossRef]
- Shao, Y.; Luo, J.; Ye, L.; Ran, H.Y.; Shi, H.M.; Zhang, C.; Wu, Q.C. Construction and Integrated Analysis of Competitive Endogenous Long Non-Coding RNA Network in Thoracic Aortic Dissection. Int J Gen Med 2021, 14, 6863–6873. [Google Scholar] [CrossRef]
- Wen, G.; An, W.; Chen, J.; Maguire, E.M.; Chen, Q.; Yang, F.; Pearce, S.W.A.; Kyriakides, M.; Zhang, L.; Ye, S.; et al. Genetic and Pharmacologic Inhibition of the Neutrophil Elastase Inhibits Experimental Atherosclerosis. J. Am. Heart Assoc. 2018, 7, e008187. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Zeng, L.; Zhang, Z.; Margariti, A.; Ali, Z.A.; Channon, K.M.; Xu, Q.; Hu, Y. Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2244–2251. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Xiao, Q.; Margariti, A.; Zhang, Z.; Zampetaki, A.; Patel, S.; Capogrossi, M.C.; Hu, Y.; Xu, Q. HDAC3 is crucial in shear- and VEGF-induced stem cell differentiation toward endothelial cells. J. Cell. Biol. 2006, 174, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Yang, F.; Guo, M.; Wen, G.; Zhang, C.; Luong le, A.; Zhu, J.; Xiao, Q.; Zhang, L. miRNA-34a reduces neointima formation through inhibiting smooth muscle cell proliferation and migration. J. Mol. Cell. Cardiol. 2015, 89, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, D.; Maguire, E.M.; He, S.; Chen, J.; An, W.; Yang, M.; Afzal, T.A.; Luong, L.A.; Zhang, L.; et al. Cbx3 inhibits vascular smooth muscle cell proliferation, migration, and neointima formation. Cardiovasc. Res. 2018, 114, 443–455. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Luong, L.A.; Bowden, N.P.; Yang, M.; Wu, W.; Zhou, X.; Liu, C.; Niu, K.; Luo, J.; Zhang, C.; et al. Cezanne is a critical regulator of pathological arterial remodelling by targeting beta-catenin signalling. Cardiovasc. Res. 2022, 118, 638–653. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Luo, Y.; Dorf, M.E.; Lionakis, M.S. Isolation of Mouse Neutrophils. Curr. Protoc. Immunol. 2015, 110, 3.20.1–3.20.15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Chen, Q.; Mei, L.; Wen, G.; An, W.; Zhou, X.; Niu, K.; Liu, C.; Ren, M.; Sun, K.; et al. Neutrophil elastase promotes neointimal hyperplasia by targeting toll-like receptor 4 (TLR4)-NF-kappaB signalling. Br. J. Pharmacol. 2021, 178, 4048–4068. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, C.; Chen, J.; Yang, M.; Afzal, T.A.; An, W.; Maguire, E.M.; He, S.; Luo, J.; Wang, X.; et al. miRNA-200c-3p promotes endothelial to mesenchymal transition and neointimal hyperplasia in artery bypass grafts. J. Pathol. 2021, 253, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Q.; An, W.; Yang, F.; Maguire, E.M.; Chen, D.; Zhang, C.; Wen, G.; Yang, M.; Dai, B.; et al. Novel Pathological Role of hnRNPA1 (Heterogeneous Nuclear Ribonucleoprotein A1) in Vascular Smooth Muscle Cell Function and Neointima Hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2182–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Chen, Q.; He, S.; Yang, M.; Maguire, E.M.; An, W.; Afzal, T.A.; Luong, L.A.; Zhang, L.; Xiao, Q. miR-22 Is a Novel Mediator of Vascular Smooth Muscle Cell Phenotypic Modulation and Neointima Formation. Circulation 2018, 137, 1824–1841. [Google Scholar] [CrossRef]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox. Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, U.; Dimagli, A.; Kaura, A.; Sinha, S.; Mariscalco, G.; Krasopoulos, G.; Moorjani, N.; Field, M.; Uday, T.; Kendal, S.; et al. Determinants of outcomes following surgery for type A acute aortic dissection: The UK National Adult Cardiac Surgical Audit. Eur. Heart J. 2021, 43, 44–52. [Google Scholar] [CrossRef]
- Kormi, I.; Nieminen, M.T.; Havulinna, A.S.; Zeller, T.; Blankenberg, S.; Tervahartiala, T.; Sorsa, T.; Salomaa, V.; Pussinen, P.J. Matrix metalloproteinase-8 and tissue inhibitor of matrix metalloproteinase-1 predict incident cardiovascular disease events and all-cause mortality in a population-based cohort. Eur. J. Prev. Cardiol. 2017, 24, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Momiyama, Y.; Ohmori, R.; Taniguchi, H.; Nakamura, H.; Ohsuzu, F. Plasma matrix metalloproteinase-8 concentrations are associated with the presence and severity of coronary artery disease. Circ. J. Off. J. Jpn. Circ. Soc. 2005, 69, 1035–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momiyama, Y.; Ohmori, R.; Tanaka, N.; Kato, R.; Taniguchi, H.; Adachi, T.; Nakamura, H.; Ohsuzu, F. High plasma levels of matrix metalloproteinase-8 in patients with unstable angina. Atherosclerosis 2010, 209, 206–210. [Google Scholar] [CrossRef]
- Jia, Y.; Guo, D.; Zhang, K.; Yang, P.; Zang, Y.; Sun, L.; Wang, Y.; Liu, F.; Shi, M.; Zhang, Y.; et al. Causal associations of serum matrix metalloproteinase-8 level with ischaemic stroke and ischaemic stroke subtypes: A Mendelian randomization study. Eur. J. Neurol. 2021, 28, 2543–2551. [Google Scholar] [CrossRef] [PubMed]
- Molloy, K.J.; Thompson, M.M.; Jones, J.L.; Schwalbe, E.C.; Bell, P.R.; Naylor, A.R.; Loftus, I.M. Unstable carotid plaques exhibit raised matrix metalloproteinase-8 activity. Circulation 2004, 110, 337–343. [Google Scholar] [CrossRef]
- van den Borne, S.W.; Cleutjens, J.P.; Hanemaaijer, R.; Creemers, E.E.; Smits, J.F.; Daemen, M.J.; Blankesteijn, W.M. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc. Pathol. 2009, 18, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Schwalbe, E.C.; Jones, J.L.; Bell, P.R.; Thompson, M.M. Matrix metalloproteinase 8 (neutrophil collagenase) in the pathogenesis of abdominal aortic aneurysm. Br. J. Surg. 2005, 92, 828–833. [Google Scholar] [CrossRef]
- Wilson, W.R.; Anderton, M.; Schwalbe, E.C.; Jones, J.L.; Furness, P.N.; Bell, P.R.; Thompson, M.M. Matrix metalloproteinase-8 and -9 are increased at the site of abdominal aortic aneurysm rupture. Circulation 2006, 113, 438–445. [Google Scholar] [CrossRef] [Green Version]
- Takagi, H.; Hari, Y.; Nakashima, K.; Kuno, T.; Ando, T. Matrix metalloproteinases and acute aortic dissection: Et Tu, Brute? Interact. Cardiovasc. Thorac. Surg. 2020, 30, 465–476. [Google Scholar] [CrossRef]
- Li, Y.; Shao, A.Z.; Jiang, H.T.; Dong, G.H.; Xu, B.; Yi, J.; Jing, H. The prominent expression of plasma matrix metalloproteinase-8 in acute thoracic aortic dissection. J. Surg. Res. 2010, 163, e99–e104. [Google Scholar] [CrossRef]
- Giachino, F.; Loiacono, M.; Lucchiari, M.; Manzo, M.; Battista, S.; Saglio, E.; Lupia, E.; Moiraghi, C.; Hirsch, E.; Mengozzi, G.; et al. Rule out of acute aortic dissection with plasma matrix metalloproteinase 8 in the emergency department. Crit. Care. 2013, 17, R33. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Du, X.M.; Jing, Q.M.; Li, X.X.; Gu, R.X.; Wang, J.; Han, Y.L. Impact of matrix metalloproteinase-8 gene variations on the risk of thoracic aortic dissection in a Chinese Han population. Mol. Biol. Rep. 2013, 40, 5953–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, T.; Shimizu-Hirota, R.; Shimoda, M.; Adachi, T.; Shimizu, H.; Weiss, S.J.; Itoh, H.; Hori, S.; Aikawa, N.; Okada, Y. Neutrophil-derived matrix metalloproteinase 9 triggers acute aortic dissection. Circulation 2012, 126, 3070–3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Liu, D.; Zhao, L.; Wang, L.; Li, Y.; Cho, K.; Tao, C.; Jiang, B. Targeted depletion of monocyte/macrophage suppresses aortic dissection with the spatial regulation of MMP-9 in the aorta. Life Sci. 2020, 254, 116927. [Google Scholar] [CrossRef]
- Trachet, B.; Piersigilli, A.; Fraga-Silva, R.A.; Aslanidou, L.; Sordet-Dessimoz, J.; Astolfo, A.; Stampanoni, M.F.; Segers, P.; Stergiopulos, N. Ascending Aortic Aneurysm in Angiotensin II-Infused Mice: Formation, Progression, and the Role of Focal Dissections. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Trachet, B.; Aslanidou, L.; Piersigilli, A.; Fraga-Silva, R.A.; Sordet-Dessimoz, J.; Villanueva-Perez, P.; Stampanoni, M.F.M.; Stergiopulos, N.; Segers, P. Angiotensin II infusion into ApoE-/- mice: A model for aortic dissection rather than abdominal aortic aneurysm? Cardiovasc. Res. 2017, 113, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Hibino, M.; Otaki, Y.; Kobeissi, E.; Pan, H.; Hibino, H.; Taddese, H.; Majeed, A.; Verma, S.; Konta, T.; Yamagata, K.; et al. Blood Pressure, Hypertension, and the Risk of Aortic Dissection Incidence and Mortality: Results From the J-SCH Study, the UK Biobank Study, and a Meta-Analysis of Cohort Studies. Circulation 2022, 145, 633–644. [Google Scholar] [CrossRef]
- Hanna, I.R.; Taniyama, Y.; Szocs, K.; Rocic, P.; Griendling, K.K. NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid. Redox Signal. 2002, 4, 899–914. [Google Scholar] [CrossRef]
- Griendling, K.K.; Ushio-Fukai, M. Reactive oxygen species as mediators of angiotensin II signaling. Regul. Pept. 2000, 91, 21–27. [Google Scholar] [CrossRef]
- Dimmeler, S.; Zeiher, A.M. Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regul. Pept. 2000, 90, 19–25. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Nazarewicz, R.R. Angiotensin II-induced production of mitochondrial reactive oxygen species: Potential mechanisms and relevance for cardiovascular disease. Antioxid. Redox Signal. 2013, 19, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Y.H.; Mohler, J.; Delafontaine, P. ANG II induces apoptosis of human vascular smooth muscle via extrinsic pathway involving inhibition of Akt phosphorylation and increased FasL expression. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2116–H2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, N.Y.; Li, J.M.; Stocker, R.; Khachigian, L.M. Angiotensin II-inducible smooth muscle cell apoptosis involves the angiotensin II type 2 receptor, GATA-6 activation, and FasL-Fas engagement. Circ. Res. 2009, 105, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, L.M.; Douglas, G.; Bendall, J.K.; McNeill, E.; Crabtree, M.J.; Hale, A.B.; Mai, A.; Li, J.M.; McAteer, M.A.; Schneider, J.E.; et al. Endothelial cell-specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation 2014, 129, 2661–2672. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Niu, K.; Ren, M.; Zhou, X.; Yang, Z.; Yang, M.; Wang, X.; Luo, J.; Shao, Y.; Zhang, C.; et al. Targeted Inhibition of Matrix Metalloproteinase-8 Prevents Aortic Dissection in a Murine Model. Cells 2022, 11, 3218. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203218
Zhang C, Niu K, Ren M, Zhou X, Yang Z, Yang M, Wang X, Luo J, Shao Y, Zhang C, et al. Targeted Inhibition of Matrix Metalloproteinase-8 Prevents Aortic Dissection in a Murine Model. Cells. 2022; 11(20):3218. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203218
Chicago/Turabian StyleZhang, Chengxin, Kaiyuan Niu, Meixia Ren, Xinmiao Zhou, Zhisheng Yang, Mei Yang, Xinxin Wang, Jun Luo, Yue Shao, Cheng Zhang, and et al. 2022. "Targeted Inhibition of Matrix Metalloproteinase-8 Prevents Aortic Dissection in a Murine Model" Cells 11, no. 20: 3218. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11203218