
Current Perspectives of Neuroendocrine Regulation in Liver Fibrosis


{kind=link}
{kind=link}
{kind=link}
Abstract
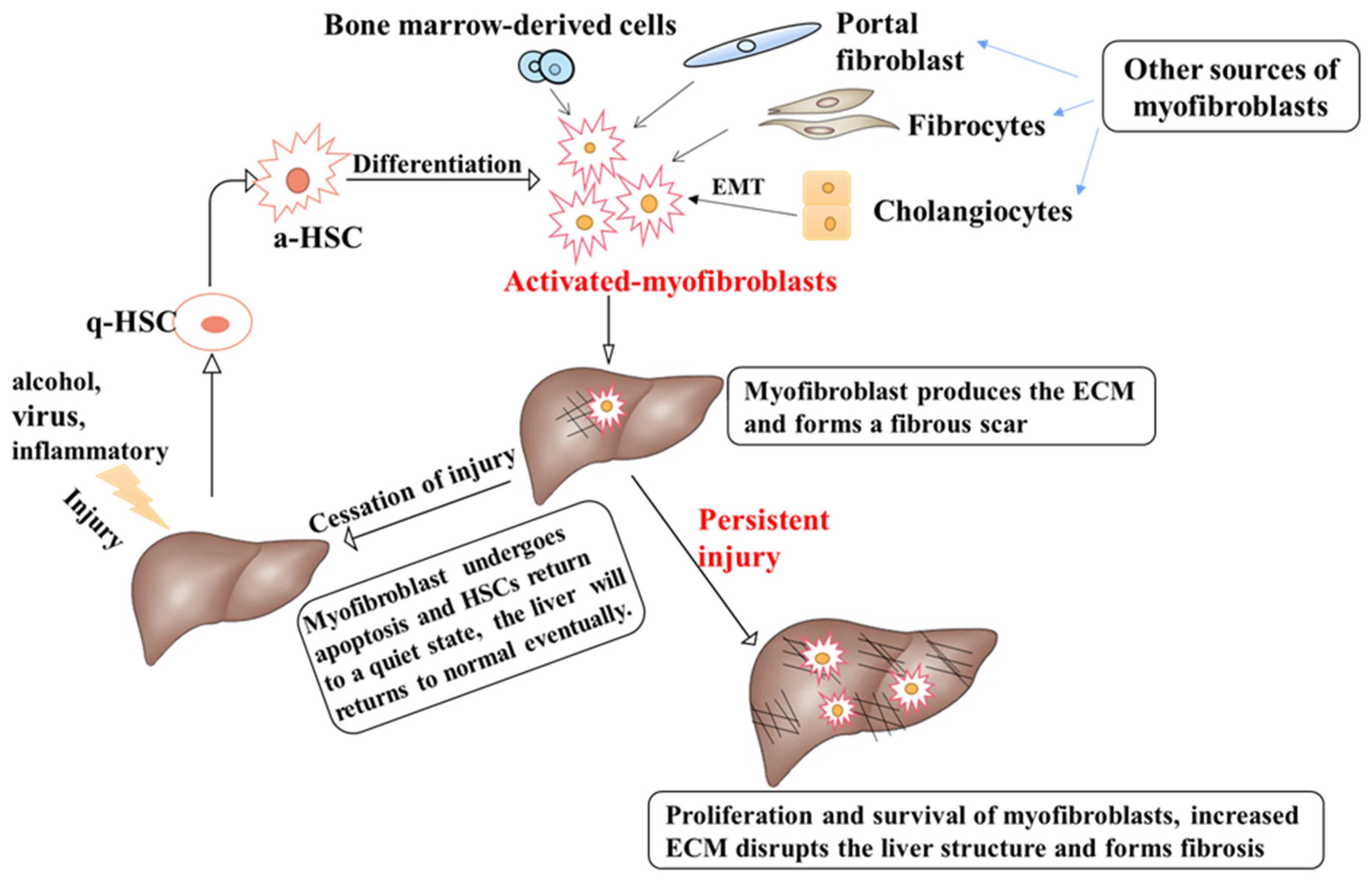
:1. Introduction
2. Pathophysiology of Liver Fibrosis Development
3. Hepatic Innervation and Neuroendocrine Compartments Are Involved in the Chronic Liver Diseases
3.1. Cholangiocytes
3.2. Hepatic Stellate Cells
3.3. Hepatic Progenitor Cells
4. The Role of Neuroendocrine Regulation during Liver Fibrosis
4.1. Renin-Angiotensin System
4.2. Cannabinoid System
4.3. Melatonin
4.4. Substance P
4.5. Serotonin
4.6. Calcitonin Gene-Related Peptide
4.7. Neuropeptide Y
4.8. Endogenous Opioid Peptides
4.9. Galanin
4.10. Secretin (Sct)
4.11. Other Neuroendocrines Involved in Liver Fibrosis
5. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.J.; Alpini, G.; Glaser, S. Hepatic nervous system and neurobiology of the liver. Compr. Physiol. 2013, 3, 655–665. [Google Scholar] [PubMed] [Green Version]
- Stoyanova, I.I.; Gulubova, M.V. Immunocytochemical study on the liver innervation in patients with cirrhosis. Acta Histochem. 2000, 102, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Parsonage, G.; Filer, A.D.; Haworth, O.; Nash, G.B.; Rainger, G.E.; Salmon, M.; Buckley, C.D. A stromal address code defined by fibroblasts. Trends Immunol. 2005, 26, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Sempowski, G.D.; Borrello, M.A.; Blieden, T.M.; Barth, R.K.; Phipps, R.P. Fibroblast heterogeneity in the healing wound. Wound Repair Regen. 1995, 3, 120–131. [Google Scholar] [CrossRef]
- Kinnman, N.; Housset, C. Peribiliary myofibroblasts in biliary type liver fibrosis. Front. Biosci. 2002, 7, d496–d503. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, A.S.; Diaz, R.; Hui, J.J.; Yanger, K.; Zong, Y.; Alpini, G.; Stanger, B.Z.; Wells, R.G. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology 2011, 53, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Scholten, D.; Osterreicher, C.H.; Scholten, A.; Iwaisako, K.; Gu, G.; Brenner, D.A.; Kisseleva, T. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology 2010, 139, 987–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benyon, R.C.; Iredale, J.P. Is liver fibrosis reversible? Gut 2000, 46, 443–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCawley, L.J.; Matrisian, L.M. Matrix metalloproteinases: They’re not just for matrix anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540. [Google Scholar] [CrossRef]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis—A systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef]
- Arthur, M.J. Fibrogenesis II. Metalloproteinases and their inhibitors in liver fibrosis. Am. J. Physiol. Liver Physiol. 2000, 279, G245–G249. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Marra, F. Hepatic stellate cells and the regulation of liver inflammation. J. Hepatol. 1999, 31, 1120–1130. [Google Scholar] [CrossRef]
- Aydin, M.M.; Akcali, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and structural features of cholangiocytes in health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallowfield, J.A.; Mizuno, M.; Kendall, T.J.; Constandinou, C.M.; Benyon, R.C.; Duffield, J.S.; Iredale, J.P. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J. Immunol. 2007, 178, 5288–5295. [Google Scholar] [CrossRef] [Green Version]
- Roskams, T.; Cassiman, D.; De Vos, R.; Libbrecht, L. Neuroregulation of the neuroendocrine compartment of the liver. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2004, 280, 910–923. [Google Scholar] [CrossRef]
- Marzioni, M.; Fava, G.; Benedetti, A. Nervous and Neuroendocrine regulation of the pathophysiology of cholestasis and of biliary carcinogenesis. World J. Gastroenterol. 2006, 12, 3471–3480. [Google Scholar] [CrossRef]
- Micera, A.; Puxeddu, I.; Lambiase, A.; Antonelli, A.; Bonini, S.; Bonini, S.; Aloe, L.; Pe’Er, J.; Levi-Schaffer, F. The pro-fibrogenic effect of nerve growth factor on conjunctival fibroblasts is mediated by transforming growth factor-beta. Clin. Exp. Allergy 2005, 35, 650–656. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Cassiman, D.; van Pelt, J.; De Vos, R.; Van Lommel, F.; Desmet, V.; Yap, S.H.; Roskams, T. Synaptophysin: A novel marker for human and rat hepatic stellate cells. Am. J. Pathol. 1999, 155, 1831–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskams, T.A.; Libbrecht, L.; Desmet, V.J. Progenitor cells in diseased human liver. Semin. Liver Dis. 2003, 23, 385–396. [Google Scholar] [PubMed]
- Roskams, T.; De Vos, R.; van den Oord, J.J.; Desmet, V. Cells with neuroendocrine features in regenerating human liver. APMIS Suppl. 1991, 23, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; Guyton, A.C.; Mizelle, H.L. Role of the renin-angiotensin system in control of sodium excretion and arterial pressure. Acta Physiol. Scand. Suppl. 1990, 591, 48–62. [Google Scholar]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 receptors: Regulation, signalling and function. Blood Press. 2003, 12, 70–88. [Google Scholar] [CrossRef]
- Santos, R.A.; Brosnihan, K.B.; Chappell, M.C.; Pesquero, J.; Chernicky, C.L.; Greene, L.J.; Ferrario, C.M. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension 1988, 11, I153–I157. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Santos, R.A.; Haibara, A.S.; Campagnole-Santos, M.J.; Simoes e Silva, A.C.; Paula, R.D.; Pinheiro, S.V.; Leite, M.F.; Lemos, V.S.; Silva, D.M.; Guerra, M.T.; et al. Characterization of a new selective antagonist for angiotensin-(1-7), D-pro7-angiotensin-(1-7). Hypertension 2003, 41, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.M.; Dos Santos, R.A.; Teixeira, M.M.; Leite, V.H.; Costa, L.P.; da Costa Dias, F.L.; Barcelos, L.S.; Collares, G.B.; Simoes e Silva, A.C. The renin-angiotensin system in a rat model of hepatic fibrosis: Evidence for a protective role of Angiotensin-(1-7). J. Hepatol. 2007, 46, 674–681. [Google Scholar] [CrossRef]
- Simões, E.; Silva, A.C.; Miranda, A.S.; Rocha, N.P.; Teixeira, A.L. Renin angiotensin system in liver diseases: Friend or foe? World J. Gastroenterol. 2017, 23, 3396. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.S. The peptide hormone angiotensin II: Its new functions in tissues and organs. Curr. Protein Pept. Sci. 2004, 5, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Gines, P.; Nicolas, J.M.; Gorbig, M.N.; Garcia-Ramallo, E.; Gasull, X.; Bosch, J.; Arroyo, V.; Rodes, J. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology 2000, 118, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Tharaux, P.L.; Chatziantoniou, C.; Fakhouri, F.; Dussaule, J.C. Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension 2000, 36, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.M.; Yang, R.Q.; Li, Y.; Ning, Z.W.; Zhang, L.L.; Zhou, G.S.; Luo, W.; Li, D.H.; Chen, Y.; Pan, M.X.; et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid. Redox Signal. 2016, 24, 795–812. [Google Scholar] [CrossRef]
- Centonze, D.; Finazzi-Agro, A.; Bernardi, G.; Maccarrone, M. The endocannabinoid system in targeting inflammatory neurodegenerative diseases. Trends Pharmacol. Sci. 2007, 28, 180–187. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Woods, S.C. Role of the endocannabinoid system in regulating cardiovascular and metabolic risk factors. Am. J. Med. 2007, 120, S19–S25. [Google Scholar] [CrossRef]
- Mallat, A.; Lotersztajn, S. Endocannabinoids as novel mediators of liver diseases. J. Endocrinol. Investig. 2006, 29, 58–65. [Google Scholar]
- Teixeira-Clerc, F.; Julien, B.; Grenard, P.; Tran, V.N.J.; Deveaux, V.; Li, L.; Serriere-Lanneau, V.; Ledent, C.; Mallat, A.; Lotersztajn, S. CB1 cannabinoid receptor antagonism: A new strategy for the treatment of liver fibrosis. Nat. Med. 2006, 12, 671–676. [Google Scholar] [CrossRef]
- Julien, B.; Grenard, P.; Teixeira-Clerc, F.; Van Nhieu, J.T.; Li, L.; Karsak, M.; Zimmer, A.; Mallat, A.; Lotersztajn, S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, S.V.; Uchinami, H.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. Anandamide induces necrosis in primary hepatic stellate cells. Hepatology 2005, 41, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Diaz-Casado, M.E.; Lima-Cabello, E.; Lopez, L.C.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef] [PubMed]
- Renzi, A.; Glaser, S.; DeMorrow, S.; Mancinelli, R.; Meng, F.; Franchitto, A.; Venter, J.; White, M.; Francis, H.; Han, Y.; et al. Melatonin inhibits cholangiocyte hyperplasia in cholestatic rats by interaction with MT1 but not MT2 melatonin receptors. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G634–G643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespo, I.; San-Miguel, B.; Fernandez, A.; Ortiz de Urbina, J.; Gonzalez-Gallego, J.; Tunon, M.J. Melatonin limits the expression of profibrogenic genes and ameliorates the progression of hepatic fibrosis in mice. Transl. Res. 2015, 165, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Shajari, S.; Laliena, A.; Heegsma, J.; Tunon, M.J.; Moshage, H.; Faber, K.N. Melatonin suppresses activation of hepatic stellate cells through RORalpha-mediated inhibition of 5-lipoxygenase. J. Pineal Res. 2015, 59, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Hong, J.M.; Lee, S.M. Melatonin enhances mitophagy and mitochondrial biogenesis in rats with carbon tetrachloride-induced liver fibrosis. J. Pineal Res. 2016, 60, 383–393. [Google Scholar] [CrossRef]
- Wu, N.; Meng, F.; Zhou, T.; Han, Y.; Kennedy, L.; Venter, J.; Francis, H.; DeMorrow, S.; Onori, P.; Invernizzi, P.; et al. Prolonged darkness reduces liver fibrosis in a mouse model of primary sclerosing cholangitis by miR-200b down-regulation. FASEB J. 2017, 31, 4305–4324. [Google Scholar] [CrossRef] [Green Version]
- Steinhoff, M.S.; von Mentzer, B.; Geppetti, P.; Pothoulakis, C.; Bunnett, N.W. Tachykinins and their receptors: Contributions to physiological control and the mechanisms of disease. Physiol. Rev. 2014, 94, 265–301. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Meng, F.; Wu, N.; Zhou, T.; Venter, J.; Francis, H.; Kennedy, L.; Glaser, T.; Bernuzzi, F.; Invernizzi, P.; et al. Substance P increases liver fibrosis by differential changes in senescence of cholangiocytes and hepatic stellate cells. Hepatology 2017, 66, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Glaser, S.; Gaudio, E.; Renzi, A.; Mancinelli, R.; Ueno, Y.; Venter, J.; White, M.; Kopriva, S.; Chiasson, V.; DeMorrow, S.; et al. Knockout of the neurokinin-1 receptor reduces cholangiocyte proliferation in bile duct-ligated mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G297–G305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Jia, X.; Zhao, J.; Cui, R.; Yan, M. Substance P promotes hepatic stellate cell proliferation and activation via the TGF-beta1/Smad-3 signaling pathway. Toxicol. Appl. Pharmacol. 2017, 329, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Jeong, J.; Jung, J.; Yoo, K.; Hong, H.S. Substance P Promotes Liver Sinusoidal Endothelium-Mediated Hepatic Regeneration by NO/HGF Regulation. J. Interferon Cytokine Res. 2019, 39, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, D.; Hannon, J.P.; Martin, G.R. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav. 2002, 71, 533–554. [Google Scholar] [CrossRef]
- Lesurtel, M.; Graf, R.; Aleil, B.; Walther, D.J.; Tian, Y.; Jochum, W.; Gachet, C.; Bader, M.; Clavien, P.A. Platelet-derived serotonin mediates liver regeneration. Science 2006, 312, 104–107. [Google Scholar] [CrossRef]
- Ruddell, R.G.; Oakley, F.; Hussain, Z.; Yeung, I.; Bryan-Lluka, L.J.; Ramm, G.A.; Mann, D.A. A role for serotonin (5-HT) in hepatic stellate cell function and liver fibrosis. Am. J. Pathol. 2006, 169, 861–876. [Google Scholar] [CrossRef] [Green Version]
- Pang, Q.; Jin, H.; Wang, Y.; Dai, M.; Liu, S.; Tan, Y.; Liu, H.; Lu, Z. Depletion of serotonin relieves concanavalin A-induced liver fibrosis in mice by inhibiting inflammation, oxidative stress, and TGF-beta1/Smads signaling pathway. Toxicol. Lett. 2021, 340, 123–132. [Google Scholar] [CrossRef]
- Polat, B.; Halici, Z.; Cadirci, E.; Karakus, E.; Bayir, Y.; Albayrak, A.; Unal, D. Liver 5-HT7 receptors: A novel regulator target of fibrosis and inflammation-induced chronic liver injury in vivo and in vitro. Int. Immunopharmacol. 2017, 43, 227–235. [Google Scholar] [CrossRef]
- Bracq, S.; Clement, B.; Pidoux, E.; Moukhtar, M.S.; Jullienne, A. CGRP is expressed in primary cultures of human hepatocytes and in normal liver. FEBS Lett. 1994, 351, 63–66. [Google Scholar] [CrossRef] [Green Version]
- Linscheid, P.; Seboek, D.; Zulewski, H.; Keller, U.; Muller, B. Autocrine/paracrine role of inflammation-mediated calcitonin gene-related peptide and adrenomedullin expression in human adipose tissue. Endocrinology 2005, 146, 2699–2708. [Google Scholar] [CrossRef]
- Barry, C.M.; Kestell, G.; Gillan, M.; Haberberger, R.V.; Gibbins, I.L. Sensory nerve fibers containing calcitonin gene-related peptide in gastrocnemius, latissimus dorsi and erector spinae muscles and thoracolumbar fascia in mice. Neuroscience 2015, 291, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Goehler, L.E.; Sternini, C. Calcitonin gene-related peptide innervation of the rat hepatobiliary system. Peptides 1996, 17, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, F.; Schifter, S.; Henriksen, J.H. Increased circulating calcitonin gene-related peptide (CGRP) in cirrhosis. J. Hepatol. 1991, 12, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Glaser, S.S.; Ueno, Y.; DeMorrow, S.; Chiasson, V.L.; Katki, K.A.; Venter, J.; Francis, H.L.; Dickerson, I.M.; DiPette, D.J.; Supowit, S.C.; et al. Knockout of alpha-calcitonin gene-related peptide reduces cholangiocyte proliferation in bile duct ligated mice. Lab. Invest. 2007, 87, 914–926. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.P.; Kim, J. Exogenous CGRP upregulates profibrogenic growth factors through PKC/JNK signaling pathway in kidney proximal tubular cells. Cell Biol. Toxicol. 2018, 34, 251–262. [Google Scholar] [CrossRef]
- Wan, Y.; Ceci, L.; Wu, N.; Zhou, T.; Chen, L.; Venter, J.; Francis, H.; Bernuzzi, F.; Invernizzi, P.; Kyritsi, K.; et al. Knockout of α-calcitonin gene-related peptide attenuates cholestatic liver injury by differentially regulating cellular senescence of hepatic stellate cells and cholangiocytes. Lab. Investig. 2019, 99, 764–776. [Google Scholar] [CrossRef]
- Inoue, N.; Magari, S.; Ito, Y.; Sakanaka, M. Distribution, possible origins and fine structure of neuropeptide Y-containing nerve fibers in the rat liver. Brain Res. 1989, 493, 87–96. [Google Scholar] [CrossRef]
- Wong, P.F.; Gall, M.G.; Bachovchin, W.W.; McCaughan, G.W.; Keane, F.M.; Gorrell, M.D. Neuropeptide Y is a physiological substrate of fibroblast activation protein: Enzyme kinetics in blood plasma and expression of Y2R and Y5R in human liver cirrhosis and hepatocellular carcinoma. Peptides 2016, 75, 80–95. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Liu, Z.W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschop, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Kim, Y.J.; Chao, P.T.; Bi, S. Overexpression of neuropeptide Y in the dorsomedial hypothalamus causes hyperphagia and obesity in rats. Obesity 2013, 21, 1086–1092. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, S.; Yu, F.; Li, H.; Guo, C.; Fan, X. Ghrelin Attenuates Liver Fibrosis through Regulation of TGF-beta1 Expression and Autophagy. Int. J. Mol. Sci. 2015, 16, 21911–21930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMorrow, S.; Onori, P.; Venter, J.; Invernizzi, P.; Frampton, G.; White, M.; Franchitto, A.; Kopriva, S.; Bernuzzi, F.; Francis, H.; et al. Neuropeptide Y inhibits cholangiocarcinoma cell growth and invasion. Am. J. Physiol. Cell Physiol. 2011, 300, C1078–C1089. [Google Scholar] [CrossRef] [PubMed]
- DeMorrow, S.; Meng, F.; Venter, J.; Leyva-Illades, D.; Francis, H.; Frampton, G.; Pae, H.Y.; Quinn, M.; Onori, P.; Glaser, S.; et al. Neuropeptide Y inhibits biliary hyperplasia of cholestatic rats by paracrine and autocrine mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G250–G257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Liu, Y.; Zhang, Y.; Sun, Y.; Sun, C.; Zhang, Y.; Lv, X. Expression of neuropeptide Y is increased in an activated human HSC cell line. Sci. Rep. 2019, 9, 9500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, D.G. 60 YEARS OF POMC: Lipotropin and beta-endorphin: A perspective. J. Mol. Endocrinol. 2016, 56, T13–T25. [Google Scholar] [CrossRef] [Green Version]
- Vaccarino, A.L.; Kastin, A.J. Endogenous opiates: 1999. Peptides 2000, 21, 1975–2034. [Google Scholar] [CrossRef]
- Mani, A.R.; Rasool, R.; Montagnese, S.; Dehpour, A.R. Endogenous opioids and liver disease. Scand. J. Gastroenterol. 2006, 41, 1–11. [Google Scholar] [CrossRef]
- Marzioni, M.; Svegliati, B.G.; Alpini, G.; Benedetti, A. Endogenous opioid peptides and chronic liver disease: From bedside to bench. J. Hepatol. 2007, 46, 583–586. [Google Scholar] [CrossRef]
- Marzioni, M.; Alpini, G.; Saccomanno, S.; de Minicis, S.; Glaser, S.; Francis, H.; Trozzi, L.; Venter, J.; Orlando, F.; Fava, G.; et al. Endogenous opioids modulate the growth of the biliary tree in the course of cholestasis. Gastroenterology 2006, 130, 1831–1847. [Google Scholar] [CrossRef]
- Bergasa, N.V.; Rothman, R.B.; Vergalla, J.; Xu, H.; Swain, M.G.; Jones, E.A. Central mu-opioid receptors are down-regulated in a rat model of cholestasis. J. Hepatol. 1992, 15, 220–224. [Google Scholar] [CrossRef]
- Bergasa, N.V.; Sabol, S.L.; Young, W.S., 3rd; Kleiner, D.E.; Jones, E.A. Cholestasis is associated with preproenkephalin mRNA expression in the adult rat liver. Am. J. Physiol. 1995, 268, G346–G354. [Google Scholar] [CrossRef] [PubMed]
- Bergasa, N.V.; Liau, S.; Homel, P.; Ghali, V. Hepatic Met-enkephalin immunoreactivity is enhanced in primary biliary cirrhosis. Liver 2002, 22, 107–113. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Candelaresi, C.; Marzioni, M.; Saccomano, S.; Roskams, T.; Casini, A.; Risaliti, A.; Salzano, R.; Cautero, N.; di Francesco, F.; et al. Role of endogenous opioids in modulating HSC activity in vitro and liver fibrosis in vivo. Gut 2008, 57, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimkhani, M.R.; Kiani, S.; Oakley, F.; Kendall, T.; Shariftabrizi, A.; Tavangar, S.M.; Moezi, L.; Payabvash, S.; Karoon, A.; Hoseininik, H.; et al. Naltrexone, an opioid receptor antagonist, attenuates liver fibrosis in bile duct ligated rats. Gut 2006, 55, 1606–1616. [Google Scholar] [CrossRef]
- Ch’Ng, J.L.; Christofides, N.D.; Anand, P.; Gibson, S.J.; Allen, Y.S.; Su, H.C.; Tatemoto, K.; Morrison, J.F.; Polak, J.M.; Bloom, S.R. Distribution of galanin immunoreactivity in the central nervous system and the responses of galanin-containing neuronal pathways to injury. Neuroscience 1985, 16, 343–354. [Google Scholar] [CrossRef]
- Smith, K.E.; Walker, M.W.; Artymyshyn, R.; Bard, J.; Borowsky, B.; Tamm, J.A.; Yao, W.J.; Vaysse, P.J.; Branchek, T.A.; Gerald, C.; et al. Cloned human and rat galanin GALR3 receptors. Pharmacology and activation of G-protein inwardly rectifying K+ channels. J. Biol. Chem. 1998, 273, 23321–23326. [Google Scholar] [CrossRef] [Green Version]
- McMillin, M.; Frampton, G.; Grant, S.; DeMorrow, S. The Neuropeptide Galanin Is Up-Regulated during Cholestasis and Contributes to Cholangiocyte Proliferation. Am. J. Pathol. 2017, 187, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Petrescu, A.D.; Grant, S.; Williams, E.; Frampton, G.; Parks, N.; Blaney, H.; Davies, M.; John, R.; Reinhart, E.H.; McMillin, M.; et al. Coordinated Targeting of Galanin Receptors on Cholangiocytes and Hepatic Stellate Cells Ameliorates Liver Fibrosis in Multidrug Resistance Protein 2 Knockout Mice. Am. J. Pathol. 2020, 190, 586–601. [Google Scholar] [CrossRef]
- He, L.; Li, Z.; Zhou, D.; Ding, Y.; Xu, L.; Chen, Y.; Fan, J. Galanin receptor 2 mediates antifibrogenic effects of galanin on hepatic stellate cells. Exp. Ther. Med. 2016, 12, 3375–3380. [Google Scholar] [CrossRef] [Green Version]
- Afroze, S.; Meng, F.; Jensen, K.; McDaniel, K.; Rahal, K.; Onori, P.; Gaudio, E.; Alpini, G.; Glaser, S.S. The physiological roles of secretin and its receptor. Ann. Transl. Med. 2013, 1, 29. [Google Scholar] [CrossRef]
- Glaser, S.; Lam, I.P.; Franchitto, A.; Gaudio, E.; Onori, P.; Chow, B.K.; Wise, C.; Kopriva, S.; Venter, J.; White, M.; et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 2010, 52, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Venter, J.; Standeford, H.; Onori, P.; Marzioni, M.; Alvaro, D.; Franchitto, A.; et al. The secretin/secretin receptor axis modulates liver fibrosis through changes in transforming growth factor-beta1 biliary secretion in mice. Hepatology 2016, 64, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Meng, F.; Zhou, T.; Venter, J.; Giang, T.K.; Kyritsi, K.; Wu, C.; Alvaro, D.; Onori, P.; Mancinelli, R.; et al. The Secretin/Secretin Receptor Axis Modulates Ductular Reaction and Liver Fibrosis through Changes in Transforming Growth Factor-beta1-Mediated Biliary Senescence. Am. J. Pathol. 2018, 188, 2264–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, H.; Meng, F.; Gaudio, E.; Alpini, G. Histamine regulation of biliary proliferation. J. Hepatol. 2012, 56, 1204–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, H.L.; Demorrow, S.; Franchitto, A.; Venter, J.K.; Mancinelli, R.A.; White, M.A.; Meng, F.; Ueno, Y.; Carpino, G.; Renzi, A.; et al. Histamine stimulates the proliferation of small and large cholangiocytes by activation of both IP3/Ca2+ and cAMP-dependent signaling mechanisms. Lab. Investig. 2012, 92, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.; Hargrove, L.; Kennedy, L.; Meng, F.; Graf-Eaton, A.; Owens, J.; Alpini, G.; Johnson, C.; Bernuzzi, F.; Demieville, J.; et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2−/− mice. Hepatology 2016, 64, 1202–1216. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.; Meadows, V.; Demieville, J.; Hargrove, L.; Virani, S.; Glaser, S.; Zhou, T.; Rinehart, E.; Jaeger, V.; Kyritsi, K.; et al. Biliary damage and liver fibrosis are ameliorated in a novel mouse model lacking l-histidine decarboxylase/histamine signaling. Lab. Investig. 2020, 100, 837–848. [Google Scholar] [CrossRef]
- de Lecea, L.; Criado, J.R.; Prospero-Garcia, O.; Gautvik, K.M.; Schweitzer, P.; Danielson, P.E.; Dunlop, C.L.; Siggins, G.R.; Henriksen, S.J.; Sutcliffe, J.G. A cortical neuropeptide with neuronal depressant and sleep-modulating properties. Nature 1996, 381, 242–245. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Chorny, A.; Robledo, G.; Delgado, M. Cortistatin, a new antiinflammatory peptide with therapeutic effect on lethal endotoxemia. J. Exp. Med. 2006, 203, 563–571. [Google Scholar] [CrossRef]
- Wang, M.; Gong, Q.; Zhang, J.; Chen, L.; Zhang, Z.; Lu, L.; Yu, D.; Han, Y.; Zhang, D.; Chen, P.; et al. Characterization of gene expression profiles in HBV-related liver fibrosis patients and identification of ITGBL1 as a key regulator of fibrogenesis. Sci. Rep. 2017, 7, 43446. [Google Scholar] [CrossRef] [Green Version]
- Benitez, R.; Caro, M.; Andres-Leon, E.; O’Valle, F.; Delgado, M. Cortistatin regulates fibrosis and myofibroblast activation in experimental hepatotoxic- and cholestatic-induced liver injury. Br. J. Pharmacol. 2022, 179, 2275–2296. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Wang, H.; Zhang, Y.; Liu, Y.; Zhou, T.; Zhou, B.; Zhang, Y.; Chen, R.; Xing, J.; He, L.; et al. Current Perspectives of Neuroendocrine Regulation in Liver Fibrosis. Cells 2022, 11, 3783. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233783
Li B, Wang H, Zhang Y, Liu Y, Zhou T, Zhou B, Zhang Y, Chen R, Xing J, He L, et al. Current Perspectives of Neuroendocrine Regulation in Liver Fibrosis. Cells. 2022; 11(23):3783. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233783
Chicago/Turabian StyleLi, Bowen, Hui Wang, Yudian Zhang, Ying Liu, Tiejun Zhou, Bingru Zhou, Ying Zhang, Rong Chen, Juan Xing, Longfei He, and et al. 2022. "Current Perspectives of Neuroendocrine Regulation in Liver Fibrosis" Cells 11, no. 23: 3783. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233783