
Intercommunication between Voltage-Gated Calcium Channels and Estrogen Receptor/Estrogen Signaling: Insights into Physiological and Pathological Conditions

Abstract
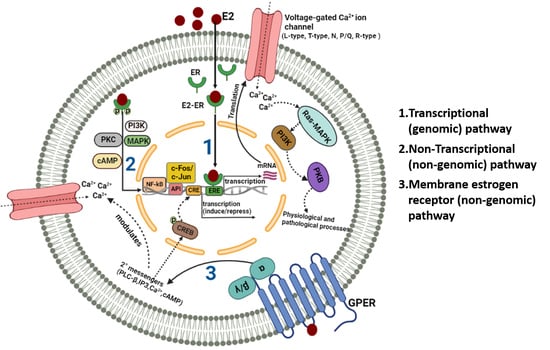
:
1. Introduction
1.1. Voltage-Gated Calcium Channels
1.2. Estrogen-Estrogen Receptor Signaling
2. Ion Channel Regulation by Estrogen
2.1. Estrogenic Regulation of VGCCs in Physiological Conditions
2.1.1. Estrogen Affects VGCC Current in Cardiovascular Tissues
Estrogen-Mediated Inhibition of VGCCs in Cardiac Tissues and Mechanisms Therein
Estrogen-Mediated Upregulation of VGCCs in Cardiac Tissues
2.1.2. Estrogen Affects VGCC Currents in Neuronal Tissues
Estrogen-Mediated Inhibition of VGCCs in Neuronal Tissues and Mechanisms Therein
Estrogen-Mediated Upregulation of VGCCs in Neuronal Tissues
2.1.3. Estrogen Modulates VGCCs in Spermatogenic Cells
2.1.4. Estrogen Modulates VGCCs in Uterine Cells
2.1.5. Estrogen Modulates VGCCs in Immune Cells
2.2. Estrogenic Regulation of VGCCs in Pathological Conditions
2.2.1. Altered Cardiac Function
2.2.2. Neurodegeneration
2.2.3. Carcinogenesis
2.2.4. Endocrine and Reproductive Defects
3. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergsman, J.B.; Wheeler, D.B.; Tsien, R.W. Classification and function of voltage-gated calcium channels. In Pharmacology of Ionic Channel Function: Activators and Inhibitors; Endo, M., Kurachi, Y., Mishina, M., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2000; Volume 147, pp. 55–85. ISBN 978-3-642-63030-9. [Google Scholar]
- Fox, A.P.; Nowycky, M.C.; Tsien, R.W. Kinetic and Pharmacological Properties Distinguishing Three Types of Calcium Currents in Chick Sensory Neurones. J. Physiol. 1987, 394, 149–172. [Google Scholar] [CrossRef] [PubMed]
- Bean, B.P. Classes of Calcium Channels in Vertebrate Cells. Annu. Rev. Physiol. 1989, 51, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Llinás, R.; Sugimori, M.; Lin, J.W.; Cherksey, B. Blocking and Isolation of a Calcium Channel from Neurons in Mammals and Cephalopods Utilizing a Toxin Fraction (FTX) from Funnel-Web Spider Poison. Proc. Natl. Acad. Sci. USA 1989, 86, 1689–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolphin, A.C. A Short History of Voltage-Gated Calcium Channels: Voltage-Gated Calcium Channels. Br. J. Pharmacol. 2006, 147, S56–S62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hell, J.W.; Westenbroek, R.E.; Warner, C.; Ahlijanian, M.K.; Prystay, W.; Gilbert, M.M.; Snutch, T.P.; Catterall, W.A. Identification and Differential Subcellular Localization of the Neuronal Class C and Class D L-Type Calcium Channel Alpha 1 Subunits. J. Cell Biol. 1993, 123, 949–962. [Google Scholar] [CrossRef]
- Melzer, W.; Herrmann-Frank, A.; Lüttgau, H.C. The Role of Ca2+ Ions in Excitation-Contraction Coupling of Skeletal Muscle Fibres. Biochim. Biophys. Acta (BBA)—Rev. Biomembr. 1995, 1241, 59–116. [Google Scholar] [CrossRef]
- Baumann, L.; Gerstner, A.; Zong, X.; Biel, M.; Wahl-Schott, C. Functional Characterization of the L-Type Ca2+ Channel Ca v 1.4α1 from Mouse Retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 708. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-Type Ca V 1.2 Calcium Channels: From In Vitro Findings to In Vivo Function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef]
- Bean, B.P. Two Kinds of Calcium Channels in Canine Atrial Cells. Differences in Kinetics, Selectivity, and Pharmacology. J. Gen. Physiol. 1985, 86, 1–30. [Google Scholar] [CrossRef]
- Mittman, S.; Guo, J.; Emerick, M.C.; Agnew, W.S. Structure and Alternative Splicing of the Gene Encoding α 1I, a Human Brain T Calcium Channel α 1 Subunit. Neurosci. Lett. 1999, 269, 121–124. [Google Scholar] [CrossRef]
- Lee, J.-H.; Daud, A.N.; Cribbs, L.L.; Lacerda, A.E.; Pereverzev, A.; Klöckner, U.; Schneider, T.; Perez-Reyes, E. Cloning and Expression of a Novel Member of the Low Voltage-Activated T-Type Calcium Channel Family. J. Neurosci. 1999, 19, 1912–1921. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, S.; Punt, E.L.; Gu, Y.; Arnoult, C.; Sakkas, D.; Barratt, C.L.R.; Publicover, S.J. Identification and Localization of T-Type Voltage-Operated Calcium Channel Subunits in Human Male Germ Cells. J. Biol. Chem. 2002, 277, 8449–8456. [Google Scholar] [CrossRef] [Green Version]
- Hillman, D.; Chen, S.; Aung, T.T.; Cherksey, B.; Sugimori, M.; Llinás, R.R. Localization of P-Type Calcium Channels in the Central Nervous System. Proc. Natl. Acad. Sci. USA 1991, 88, 7076–7080. [Google Scholar] [CrossRef] [Green Version]
- Mintz, I.M.; Adams, M.E.; Bean, B.P. P-Type Calcium Channels in Rat Central and Peripheral Neurons. Neuron 1992, 9, 85–95. [Google Scholar] [CrossRef]
- Vajna, R.; Schramm, M.; Pereverzev, A.; Arnhold, S.; Grabsch, H.; Klockner, U.; Perez-Reyes, E.; Hescheler, J.; Schneider, T. New Isoform of the Neuronal Ca2+ Channel Alpha1E Subunit in Islets of Langerhans and Kidney. Distribution of Voltage-Gated Ca2+ Channel Alpha1 Subunits in Cell Lines and Tissues. Eur. J. Biochem. 1998, 257, 274–285. [Google Scholar] [CrossRef]
- Moosmang, S. Dominant Role of Smooth Muscle L-Type Calcium Channel Cav1.2 for Blood Pressure Regulation. EMBO J. 2003, 22, 6027–6034. [Google Scholar] [CrossRef]
- Striessnig, J.; Pinggera, A.; Kaur, G.; Bock, G.; Tuluc, P. L-type Ca2+ Channels in Heart and Brain. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2014, 3, 15–38. [Google Scholar] [CrossRef]
- Xu, M.; Welling, A.; Paparisto, S.; Hofmann, F.; Klugbauer, N. Enhanced Expression of L-Type Cav1.3 Calcium Channels in Murine Embryonic Hearts from Cav1.2-Deficient Mice. J. Biol. Chem. 2003, 278, 40837–40841. [Google Scholar] [CrossRef]
- Platzer, J.; Engel, J.; Schrott-Fischer, A.; Stephan, K.; Bova, S.; Chen, H.; Zheng, H.; Striessnig, J. Congenital Deafness and Sinoatrial Node Dysfunction in Mice Lacking Class D L-Type Ca2+ Channels. Cell 2000, 102, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Lingle, C.J.; Martinez-Espinosa, P.L.; Guarina, L.; Carbone, E. Roles of Na+, Ca2+, and K+ Channels in the Generation of Repetitive Firing and Rhythmic Bursting in Adrenal Chromaffin Cells. Pflug. Arch.-Eur. J. Physiol. 2018, 470, 39–52. [Google Scholar] [CrossRef]
- Eguchi, K.; Montanaro, J.; Le Monnier, E.; Shigemoto, R. The Number and Distinct Clustering Patterns of Voltage-Gated Calcium Channels in Nerve Terminals. Front. Neuroanat. 2022, 16, 846615. [Google Scholar] [CrossRef]
- Monteil, A.; Chemin, J.; Bourinet, E.; Mennessier, G.; Lory, P.; Nargeot, J. Molecular and Functional Properties of the Human A1G Subunit That Forms T-Type Calcium Channels. J. Biol. Chem. 2000, 275, 6090–6100. [Google Scholar] [CrossRef] [Green Version]
- Perez-Reyes, E. Molecular Physiology of Low-Voltage-Activated T-Type Calcium Channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, L.L.; Lee, J.-H.; Yang, J.; Satin, J.; Zhang, Y.; Daud, A.; Barclay, J.; Williamson, M.P.; Fox, M.; Rees, M.; et al. Cloning and Characterization of A1H From Human Heart, a Member of the T-Type Ca2+ Channel Gene Family. Circ. Res. 1998, 83, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.E.; Washburn, M.S.; Hans, M.; Urrutia, A.; Brust, P.F.; Prodanovich, P.; Harpold, M.M.; Stauderman, K.A. Structure and Functional Characterization of a Novel Human Low-Voltage Activated Calcium Channel. J. Neurochem. 1999, 72, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Perez-Reyes, E.; Cribbs, L.L.; Daud, A.; Lacerda, A.E.; Barclay, J.; Williamson, M.P.; Fox, M.; Rees, M.; Lee, J.-H. Molecular Characterization of a Neuronal Low-Voltage-Activated T-Type Calcium Channel. Nature 1998, 391, 896–900. [Google Scholar] [CrossRef]
- García-Delgado, N.; Velasco, M.; Sánchez-Soto, C.; Díaz-García, C.M.; Hiriart, M. Calcium Channels in Postnatal Development of Rat Pancreatic Beta Cells and Their Role in Insulin Secretion. Front. Endocrinol. 2018, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Dolphin, A.C. Voltage-Gated Calcium Channels and Their Auxiliary Subunits: Physiology and Pathophysiology and Pharmacology: Voltage-Gated Calcium Channels. J. Physiol. 2016, 594, 5369–5390. [Google Scholar] [CrossRef]
- Lanzetti, S.; Di Biase, V. Small Molecules as Modulators of Voltage-Gated Calcium Channels in Neurological Disorders: State of the Art and Perspectives. Molecules 2022, 27, 1312. [Google Scholar] [CrossRef]
- Sandoval, A.; Duran, P.; Corzo-López, A.; Fernández-Gallardo, M.; Muñoz-Herrera, D.; Leyva-Leyva, M.; González-Ramírez, R.; Felix, R. The Role of Voltage-Gated Calcium Channels in the Pathogenesis of Parkinson’s Disease. Int. J. Neurosci. 2022, 1–21. [Google Scholar] [CrossRef]
- Luebke, J.I.; Dunlap, K.; Turner, T.J. Multiple Calcium Channel Types Control Glutamatergic Synaptic Transmission in the Hippocampus. Neuron 1993, 11, 895–902. [Google Scholar] [CrossRef]
- Fox, A.P.; Cahill, A.L.; Currie, K.P.M.; Grabner, C.; Harkins, A.B.; Herring, B.; Hurley, J.H.; Xie, Z. N- and P/Q-Type Ca2+ Channels in Adrenal Chromaffin Cells: N- and P/Q-Type Ca2+ Channels. Acta Physiol. 2007, 192, 247–261. [Google Scholar] [CrossRef]
- Hirning, L.D.; Fox, A.P.; McCleskey, E.W.; Olivera, B.M.; Thayer, S.A.; Miller, R.J.; Tsien, R.W. Dominant Role of N-Type Ca2+ Channels in Evoked Release of Norepinephrine from Sympathetic Neurons. Science 1988, 239, 57–61. [Google Scholar] [CrossRef]
- Altier, C.; Dale, C.S.; Kisilevsky, A.E.; Chapman, K.; Castiglioni, A.J.; Matthews, E.A.; Evans, R.M.; Dickenson, A.H.; Lipscombe, D.; Vergnolle, N.; et al. Differential Role of N-Type Calcium Channel Splice Isoforms in Pain. J. Neurosci. 2007, 27, 6363–6373. [Google Scholar] [CrossRef] [Green Version]
- Price, T.J.; Rashid, M.H.; Millecamps, M.; Sanoja, R.; Entrena, J.M.; Cervero, F. Decreased Nociceptive Sensitization in Mice Lacking the Fragile X Mental Retardation Protein: Role of MGluR1/5 and MTOR. J. Neurosci. 2007, 27, 13958–13967. [Google Scholar] [CrossRef] [Green Version]
- Weiergräber, M.; Pereverzev, A.; Vajna, R.; Henry, M.; Schramm, M.; Nastainczyk, W.; Grabsch, H.; Schneider, T. Immunodetection of A1E Voltage-Gated Ca2+ Channel in Chromogranin-Positive Muscle Cells of Rat Heart, and in Distal Tubules of Human Kidney. J. Histochem. Cytochem. 2000, 48, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Kamp, M.A.; Krieger, A.; Henry, M.; Hescheler, J.; Weiergräber, M.; Schneider, T. Presynaptic ‘Ca v 2.3-Containing’ E-Type Ca2+ Channels Share Dual Roles during Neurotransmitter Release. Eur. J. Neurosci. 2005, 21, 1617–1625. [Google Scholar] [CrossRef]
- Karaki, H.; Ozaki, H.; Hori, M.; Mitsui-Saito, M.; Amano, K.; Harada, K.; Miyamoto, S.; Nakazawa, H.; Won, K.J.; Sato, K. Calcium Movements, Distribution, and Functions in Smooth Muscle. Pharmacol. Rev. 1997, 49, 157–230. [Google Scholar]
- Mangoni, M.E.; Couette, B.; Bourinet, E.; Platzer, J.; Reimer, D.; Striessnig, J.; Nargeot, J. Functional Role of L-Type Ca v 1.3 Ca2+ Channels in Cardiac Pacemaker Activity. Proc. Natl. Acad. Sci. USA 2003, 100, 5543–5548. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, N.; Irisawa, H.; Kameyama, M. Contribution of Two Types of Calcium Currents to the Pacemaker Potentials of Rabbit Sino-Atrial Node Cells. J. Physiol. 1988, 395, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Talavera, K.; Verkhratsky, A. T-Type Calcium Channels: The Never Ending Story. Cell Calcium 2006, 40, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Carbone, E.; Lux, H.D. A Low Voltage-Activated Calcium Conductance in Embryonic Chick Sensory Neurons. Biophys. J. 1984, 46, 413–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemin, J.; Monteil, A.; Perez-Reyes, E.; Bourinet, E.; Nargeot, J.; Lory, P. Specific Contribution of Human T-type Calcium Channel Isotypes (α1G, α 1H and α 1I) to Neuronal Excitability. J. Physiol. 2002, 540, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Mochizuki, T.; Xie, J.; Fischler, W.; Manger, J.P.; Talley, E.M.; Scammell, T.E.; Tonegawa, S. Thalamic Ca v 3.1 T-Type Ca 2 + Channel Plays a Crucial Role in Stabilizing Sleep. Proc. Natl. Acad. Sci. USA 2005, 102, 1743–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, S.M.; Snutch, T.P. Contributions of T-Type Calcium Channel Isoforms to Neuronal Firing. Channels 2010, 4, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Ophoff, R.A.; Terwindt, G.M.; Frants, R.R.; Ferrari, M.D. P/Q-Type Ca2+ Channel Defects in Migraine, Ataxia and Epilepsy. Trends Pharmacol. Sci. 1998, 19, 121–127. [Google Scholar] [CrossRef]
- Rajakulendran, S.; Graves, T.D.; Labrum, R.W.; Kotzadimitriou, D.; Eunson, L.; Davis, M.B.; Davies, R.; Wood, N.W.; Kullmann, D.M.; Hanna, M.G.; et al. Genetic and Functional Characterisation of the P/Q Calcium Channel in Episodic Ataxia with Epilepsy: Variation of CACNA1A in Episodic Ataxia and Epilepsy. J. Physiol. 2010, 588, 1905–1913. [Google Scholar] [CrossRef]
- Mezler, M.; Barghorn, S.; Schoemaker, H.; Gross, G.; Nimmrich, V. A β-Amyloid Oligomer Directly Modulates P/Q-Type Calcium Currents in Xenopus Oocytes: Aβ Oligomers Modulate P/Q-Type Calcium Currents. Br. J. Pharmacol. 2012, 165, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Boyle, L.; Kaufmann, W.E. The Behavioral Phenotype of FMR1 Mutations. Am. J. Med. Genet. 2010, 154C, 469–476. [Google Scholar] [CrossRef]
- Wormuth, C.; Lundt, A.; Henseler, C.; Müller, R.; Broich, K.; Papazoglou, A.; Weiergräber, M. Review: Cav2.3 R-Type Voltage-Gated Ca2+ Channels—Functional Implications in Convulsive and Non-Convulsive Seizure Activity. Open Neurol. J. 2016, 10, 99–126. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.; Neumaier, F.; Hescheler, J.; Alpdogan, S. Cav2.3 R-Type Calcium Channels: From Its Discovery to Pathogenic de Novo CACNA1E Variants: A Historical Perspective. Pflug. Arch.-Eur. J. Physiol. 2020, 472, 811–816. [Google Scholar] [CrossRef]
- Striessnig, J.; Bolz, H.J.; Koschak, A. Channelopathies in Cav1.1, Cav1.3, and Cav1.4 Voltage-Gated L-Type Ca2+ Channels. Pflug. Arch.-Eur. J. Physiol. 2010, 460, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Soong, T.W. CaV1.2 Channelopathies: From Arrhythmias to Autism, Bipolar Disorder, and Immunodeficiency. Pflug. Arch.-Eur. J. Physiol. 2010, 460, 353–359. [Google Scholar] [CrossRef]
- Powell, K.L.; Cain, S.M.; Snutch, T.P.; O’Brien, T.J. Low Threshold T-Type Calcium Channels as Targets for Novel Epilepsy Treatments: T-Channels and Epilepsy Treatments. Br. J. Clin. Pharmacol. 2014, 77, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Weiss, N.; Zamponi, G.W. Genetic T-Type Calcium Channelopathies. J. Med. Genet. 2020, 57, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lory, P.; Nicole, S.; Monteil, A. Neuronal Cav3 Channelopathies: Recent Progress and Perspectives. Pflug. Arch.-Eur. J. Physiol. 2020, 472, 831–844. [Google Scholar] [CrossRef]
- Rao, V.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-Gated Ion Channels in Cancer Cell Proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef]
- Phan, N.N.; Wang, C.-Y.; Chen, C.-F.; Sun, Z.; Lai, M.-D.; Lin, Y.-C. Voltage-Gated Calcium Channels: Novel Targets for Cancer Therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Saha, S. T-Type Voltage Gated Calcium Channels: A Target in Breast Cancer? Breast Cancer Res. Treat. 2019, 173, 11–21. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.T.; Huang, L.; Pottle, J.E.; Liu, K.; Yang, Y.; Zeng, X.; Keyser, B.M.; Agrawal, K.C.; Hansen, J.B.; Li, M. Selective Blockade of T-Type Ca2+ Channels Suppresses Human Breast Cancer Cell Proliferation. Cancer Lett. 2008, 267, 116–124. [Google Scholar] [CrossRef]
- Ohkubo, T.; Yamazaki, J. T-Type Voltage-Activated Calcium Channel Cav3.1, but Not Cav3.2, IsInvolved in the Inhibition of Proliferation and Apoptosis in MCF-7 Human BreastCancer Cells. Int. J. Oncol. 2012, 41, 267–275. [Google Scholar] [CrossRef]
- Jacquemet, G.; Baghirov, H.; Georgiadou, M.; Sihto, H.; Peuhu, E.; Cettour-Janet, P.; He, T.; Perälä, M.; Kronqvist, P.; Joensuu, H.; et al. L-Type Calcium Channels Regulate Filopodia Stability and Cancer Cell Invasion Downstream of Integrin Signalling. Nat. Commun. 2016, 7, 13297. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, N.; Carmine-Simmen, K.; Nair, R.; Wang, C.; Moghadas-Jafari, S.; Blaser, H.; Tran-Thanh, D.; Wang, D.; Wang, P.; Wang, J.; et al. Amplification of a Calcium Channel Subunit CACNG4 Increases Breast Cancer Metastasis. EBioMedicine 2020, 52, 102646. [Google Scholar] [CrossRef] [Green Version]
- Simpson, E.R.; Misso, M.; Hewitt, K.N.; Hill, R.A.; Boon, W.C.; Jones, M.E.; Kovacic, A.; Zhou, J.; Clyne, C.D. Estrogen—The Good, the Bad, and the Unexpected. Endocr. Rev. 2005, 26, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Koos, R.D. Minireview: Putting Physiology Back into Estrogens’ Mechanism of Action. Endocrinology 2011, 152, 4481–4488. [Google Scholar] [CrossRef] [Green Version]
- Leygue, E.; Gol-Winkler, R.; Gompel, A.; Louis-Sylvestre, C.; Soquet, L.; Staub, S.; Kuttenn, F.; Mauvais-Jarvis, P. Estradiol Stimulates C-Myc Proto-Oncogene Expression in Normal Human Breast Epithelial Cells in Culture. J. Steroid Biochem. Mol. Biol. 1995, 52, 299–305. [Google Scholar] [CrossRef]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen Accelerates the Resolution of Inflammation in Macrophagic Cells. Sci. Rep. 2015, 5, 15224. [Google Scholar] [CrossRef] [Green Version]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular Mechanism of Estrogen-Estrogen Receptor Signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Gong, X.; Yang, X.; Shang, X.; Du, Q.; Liao, Q.; Xie, R.; Chen, Y.; Xu, J. The Roles of Estrogen and Estrogen Receptors in Gastrointestinal Disease. Oncol. Lett. 2019, 18, 5673–5680. [Google Scholar] [CrossRef] [Green Version]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen Receptors Alpha (ERα) and Beta (ERβ): Subtype-Selective Ligands and Clinical Potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Piperigkou, Z.; Karamanos, N.K. Estrogen Receptor-Mediated Targeting of the Extracellular Matrix Network in Cancer. Semin. Cancer Biol. 2020, 62, 116–124. [Google Scholar] [CrossRef]
- Thomas, C.; Gustafsson, J.-Å. The Different Roles of ER Subtypes in Cancer Biology and Therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Pettersson, K.; Delaunay, F.; Gustafsson, J.-Å. Estrogen Receptor β Acts as a Dominant Regulator of Estrogen Signaling. Oncogene 2000, 19, 4970–4978. [Google Scholar] [CrossRef] [Green Version]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor Beta (ERβ): A Ligand Activated Tumor Suppressor. Front. Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef]
- Arnal, J.-F.; Lenfant, F.; Metivier, R.; Flouriot, G.; Henrion, D.; Adlanmerini, M.; Fontaine, C.; Gourdy, P.; Chambon, P.; Katzenellenbogen, B.; et al. Membrane and Nuclear Estrogen Receptor Alpha Actions: From Tissue Specificity to Medical Implications. Physiol. Rev. 2017, 97, 1045–1087. [Google Scholar] [CrossRef]
- Rusidzé, M.; Adlanmérini, M.; Chantalat, E.; Raymond-Letron, I.; Cayre, S.; Arnal, J.-F.; Deugnier, M.-A.; Lenfant, F. Estrogen Receptor-α Signaling in Post-Natal Mammary Development and Breast Cancers. Cell. Mol. Life Sci. 2021, 78, 5681–5705. [Google Scholar] [CrossRef]
- Burns, K.A.; Korach, K.S. Estrogen Receptors and Human Disease: An Update. Arch. Toxicol. 2012, 86, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Faltas, C.L.; LeBron, K.A.; Holz, M.K. Unconventional Estrogen Signaling in Health and Disease. Endocrinology 2020, 161, bqaa030. [Google Scholar] [CrossRef] [PubMed]
- Rajan, A.; Nadhan, R.; Latha, N.R.; Krishnan, N.; Warrier, A.V.; Srinivas, P. Deregulated Estrogen Receptor Signaling and DNA Damage Response in Breast Tumorigenesis. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2021, 1875, 188482. [Google Scholar] [CrossRef]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast Cancer in Young Women: An Overview. Updates Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef]
- Zubair, M.; Wang, S.; Ali, N. Advanced Approaches to Breast Cancer Classification and Diagnosis. Front. Pharmacol. 2021, 11, 632079. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Ariazi, J.L.; Cordera, F.; Jordan, V.C. Estrogen Receptors as Therapeutic Targets in Breast Cancer. Curr. Top. Med. Chem. 2006, 6, 181–202. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective Estrogen Receptor Modulators (SERMs) and Selective Estrogen Receptor Degraders (SERDs) in Cancer Treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Lainé, M.; Fanning, S.W.; Chang, Y.-F.; Green, B.; Greene, M.E.; Komm, B.; Kurleto, J.D.; Phung, L.; Greene, G.L. Lasofoxifene as a Potential Treatment for Therapy-Resistant ER-Positive Metastatic Breast Cancer. Breast Cancer Res. 2021, 23, 54. [Google Scholar] [CrossRef]
- Leygue, E.; Murphy, L.C. A Bi-Faceted Role of Estrogen Receptor β in Breast Cancer. Endocr.-Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef]
- Häring, J.; Schüler, S.; Lattrich, C.; Ortmann, O.; Treeck, O. Role of Estrogen Receptor β in Gynecological Cancer. Gynecol. Oncol. 2012, 127, 673–676. [Google Scholar] [CrossRef]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal Growth Factor Receptor as a Potential Therapeutic Target in Triple-Negative Breast Cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Lev, S. Targeted Therapy and Drug Resistance in Triple-Negative Breast Cancer: The EGFR Axis. Biochem. Soc. Trans. 2020, 48, 657–665. [Google Scholar] [CrossRef] [PubMed]
- You, K.S.; Yi, Y.W.; Cho, J.; Park, J.-S.; Seong, Y.-S. Potentiating Therapeutic Effects of Epidermal Growth Factor Receptor Inhibition in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 589. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.K.; Oturkar, C.C.; Adams, C.; Wickramasekera, N.; Bansal, S.; Medisetty, R.; Miller, A.; Swetzig, W.M.; Silwal-Pandit, L.; Børresen-Dale, A.-L.; et al. TP53 Status as a Determinant of Pro- vs Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 1202–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, N.; Silveyra, P. Estrogen Receptor Signaling Mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Wellman, G.C.; Brayden, J.E.; Nelson, M.T. A Proposed mechanism for the cardioprotective effect of oestrogen in women: Enhanced endothelial nitric oxide release decreases coronary artery reactivity. Clin. Exp. Pharmacol. Physiol. 1996, 23, 260–266. [Google Scholar] [CrossRef]
- Ritchie, A.K. Estrogen Increases Low Voltage-Activated Calcium Current Density in GH3 Anterior Pituitary Cells. Endocrinology 1993, 132, 1621–1629. [Google Scholar] [CrossRef]
- Joels, M.; Karst, H. Effects of Estradiol and Progesterone on Voltage-Gated Calcium and Potassium Conductances in Rat CA1 Hippocampal Neurons. J. Neurosci. 1995, 15, 4289–4297. [Google Scholar] [CrossRef]
- Nascimento, D.S.; Reis, C.U.; Goldenberg, R.C.; Ortiga-Carvalho, T.M.; Pazos-Moura, C.C.; Guggino, S.E.; Guggino, W.B.; Morales, M.M. Estrogen Modulates ClC-2 Chloride Channel Gene Expression in Rat Kidney. Pflug. Arch.-Eur. J. Physiol. 2003, 446, 593–599. [Google Scholar] [CrossRef]
- Kow, L.-M.; Pfaff, D.W. Rapid Estrogen Actions on Ion Channels: A Survey in Search for Mechanisms. Steroids 2016, 111, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Restrepo-Angulo, I.; Bañuelos, C.; Camacho, J. Ion Channel Regulation by Sex Steroid Hormones and Vitamin D in Cancer: A Potential Opportunity for Cancer Diagnosis and Therapy. Front. Pharmacol. 2020, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Machuki, J.O.; Wu, Q.; Shi, M.; Fu, L.; Adekunle, A.O.; Tao, X.; Xu, C.; Hu, X.; Yin, Z.; et al. Estrogen and Calcium Handling Proteins: New Discoveries and Mechanisms in Cardiovascular Diseases. Am. J. Physiol.-Heart Circ. Physiol. 2020, 318, H820–H829. [Google Scholar] [CrossRef] [PubMed]
- Batra, S.; Bengtsson, B. Effects of Diethylstilboestrol and Ovarian Steroids on the Contractile Responses and Calcium Movements in Rat Uterine Smooth Muscle. J. Physiol. 1978, 276, 329–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, D.R.; Coulson, P.B. Estrogen Receptors and Effects of Estrogen on Membrane Electrical Properties of Coronary Vascular Smooth Muscle. J. Cell. Physiol. 1979, 100, 375–382. [Google Scholar] [CrossRef]
- Shan, J.; Resnick, L.M.; Liu, Q.Y.; Wu, X.C.; Barbagallo, M.; Pang, P.K. Vascular Effects of 17 Beta-Estradiol in Male Sprague-Dawley Rats. Am. J. Physiol.-Heart Circ. Physiol. 1994, 266, H967–H973. [Google Scholar] [CrossRef]
- Jiang, C.; Poole-Wilson, P.A.; Sarrel, P.M.; Mochizuki, S.; Collins, P.; MacLeod, K.T. Effect of 17β-Oestradiol on Contraction, Ca2+ Current and Intracellular Free Ca2+ in Guinea-Pig Isolated Cardiac Myocytes. Br. J. Pharmacol. 1992, 106, 739–745. [Google Scholar] [CrossRef]
- Grohe, C.; Kahlert, S.; Löbbert, K.; Meyer, R.; Linz, K.W.; Karas, R.H.; Vetter, H. Modulation of Hypertensive Heart Disease by Estrogen. Steroids 1996, 61, 201–204. [Google Scholar] [CrossRef]
- Meyer, R.; Linz, K.W.; Surges, R.; Meinardus, S.; Vees, J.; Hoffmann, A.; Windholz, O.; Grohé, C. Rapid Modulation of L-Type Calcium Current by Acutely Applied Oestrogens in Isolated Cardiac Myocytes from Human, Guinea-Pig and Rat. Exp. Physiol. 1998, 83, 305–321. [Google Scholar] [CrossRef] [Green Version]
- Berger, F.; Borchard, U.; Hafner, D.; Pütz, I.; Weis, T.M. Effects of 17β-Estradiol on Action Potentials and Ionic Currents in Male Rat Ventricular Myocytes. Naunyn-Schmiedeberg’s Arch Pharm. 1997, 356, 788–796. [Google Scholar] [CrossRef]
- Cairrão, E.; Alvarez, E.; Carvas, J.M.; Santos-Silva, A.J.; Verde, I. Non-Genomic Vasorelaxant Effects of 17β-Estradiol and Progesterone in Rat Aorta Are Mediated by L-Type Ca2+ Current Inhibition. Acta Pharmacol. Sin. 2012, 33, 615–624. [Google Scholar] [CrossRef]
- Jiang, C.; Sarrel, P.M.; Poole-Wilson, P.A.; Collins, P. Acute Effect of 17 Beta-Estradiol on Rabbit Coronary Artery Contractile Responses to Endothelin-1. Am. J. Physiol.-Heart Circ. Physiol. 1992, 263, H271–H275. [Google Scholar] [CrossRef] [PubMed]
- Ogata, R.; Inoue, Y.; Nakano, H.; Ito, Y.; Kitamura, K. Oestradiol-Induced Relaxation of Rabbit Basilar Artery by Inhibition of Voltage-Dependent Ca Channels through GTP-Binding Protein. Br. J. Pharmacol. 1996, 117, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitazawa, T.; Hamada, E.; Kitazawa, K.; Gaznabi, A.K. Non-Genomic Mechanism of 17 Beta-Oestradiol-Induced Inhibition of Contraction in Mammalian Vascular Smooth Muscle. J. Physiol. 1997, 499, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Salom, J.B.; Burguete, M.C.; Pérez-Asensio, F.J.; Centeno, J.M.; Torregrosa, G.; Alborch, E. Acute Relaxant Effects of 17-β-Estradiol through Non-Genomic Mechanisms in Rabbit Carotid Artery. Steroids 2002, 67, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Pugach, E.K.; Blenck, C.L.; Dragavon, J.M.; Langer, S.J.; Leinwand, L.A. Estrogen Receptor Profiling and Activity in Cardiac Myocytes. Mol. Cell. Endocrinol. 2016, 431, 62–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, R.D.; Sperti, G.; Colucci, W.S.; Clapham, D.E. Phorbol Ester Increases the Dihydropyridine-Sensitive Calcium Conductance in a Vascular Smooth Muscle Cell Line. Circ. Res. 1988, 62, 1049–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Renterghem, C.; Romey, G.; Lazdunski, M. Vasopressin Modulates the Spontaneous Electrical Activity in Aortic Cells (Line A7r5) by Acting on Three Different Types of Ionic Channels. Proc. Natl. Acad. Sci. USA 1988, 85, 9365–9369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, R.T.; Cohen, C.J. Nimodipine Block of Calcium Channels in Rat Vascular Smooth Muscle Cell Lines. Exceptionally High-Affinity Binding in A7r5 and A10 Cells. J. Gen. Physiol. 1989, 94, 669–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Ram, J.L.; Standley, P.R.; Sowers, J.R. 17 Beta-Estradiol Attenuates Voltage-Dependent Ca2+ Currents in A7r5 Vascular Smooth Muscle Cell Line. Am. J. Physiol.-Cell Physiol. 1994, 266, C975–C980. [Google Scholar] [CrossRef]
- Ullrich, N.D.; Koschak, A.; MacLeod, K.T. Oestrogen Directly Inhibits the Cardiovascular L-Type Ca2+ Channel Cav1.2. Biochem. Biophys. Res. Commun. 2007, 361, 522–527. [Google Scholar] [CrossRef]
- Holm, A.; Hellstrand, P.; Olde, B.; Svensson, D.; Leeb-Lundberg, L.M.F.; Nilsson, B.-O. The G Protein-Coupled Estrogen Receptor 1 (GPER1/GPR30) Agonist G-1 Regulates Vascular Smooth Muscle Cell Ca2+ Handling. J. Vasc. Res. 2013, 50, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, V.; Wauson, E.; Christian, D.; Clayton, S.; Giles, J.; Tran, Q.-K. Regulation of Beta Adrenoceptor-Mediated Myocardial Contraction and Calcium Dynamics by the G Protein-Coupled Estrogen Receptor 1. Biochem. Pharmacol. 2020, 171, 113727. [Google Scholar] [CrossRef] [PubMed]
- Marni, F.; Wang, Y.; Morishima, M.; Shimaoka, T.; Uchino, T.; Zheng, M.; Kaku, T.; Ono, K. 17 Beta-Estradiol Modulates Expression of Low-Voltage-Activated Ca(V)3.2 T-Type Calcium Channel via Extracellularly Regulated Kinase Pathway in Cardiomyocytes. Endocrinology 2009, 150, 879–888. [Google Scholar] [CrossRef]
- Johnson, B.D.; Zheng, W.; Korach, K.S.; Scheuer, T.; Catterall, W.A.; Rubanyi, G.M. Increased Expression of the Cardiac L-Type Calcium Channel in Estrogen Receptor–Deficient Mice. J. Gen. Physiol. 1997, 110, 135–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.J.F.; Dalton, R.J.; Joseph, B.K.; Thakali, K.M.; Rusch, N.J. 17 β -Estradiol Reduces Cav1.2 Channel Abundance and Attenuates Ca2+-Dependent Contractions in Coronary Arteries. Pharmacol. Res. Perspect. 2017, 5, e00358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Chen, G.; Papp, R.; DeFranco, D.B.; Zeng, F.; Salama, G. Oestrogen Upregulates L-Type Ca2+ Channels via Oestrogen-Receptor-α by a Regional Genomic Mechanism in Female Rabbit Hearts. J. Physiol. 2012, 590, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Yang, X.; Alber, S.; Shusterman, V.; Salama, G. Regional Genomic Regulation of Cardiac Sodium-Calcium Exchanger by Oestrogen: Regional Genomic Regulation of NCX by Oestrogen. J. Physiol. 2011, 589, 1061–1080. [Google Scholar] [CrossRef] [PubMed]
- Papp, R.; Bett, G.C.L.; Lis, A.; Rasmusson, R.L.; Baczkó, I.; Varró, A.; Salama, G. Genomic Upregulation of Cardiac Cav1.2α and NCX1 by Estrogen in Women. Biol. Sex Differ. 2017, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Mao, X.; Xu, G.; Xing, S.; Chattopadhyay, A.; Jin, S.; Salama, G. Estradiol Up-Regulates L-Type Ca 2+ Channels via Membrane-Bound Estrogen Receptor/Phosphoinositide-3-Kinase/Akt/CAMP Response Element-Binding Protein Signaling Pathway. Heart Rhythm 2018, 15, 741–749. [Google Scholar] [CrossRef]
- Machuki, J.O.; Zhang, H.-Y.; Geng, J.; Fu, L.; Adzika, G.K.; Wu, L.; Shang, W.; Wu, J.; Kexue, L.; Zhao, Z.; et al. Estrogen Regulation of Cardiac CAMP-L-Type Ca2+ Channel Pathway Modulates Sex Differences in Basal Contraction and Responses to Β2AR-Mediated Stress in Left Ventricular Apical Myocytes. Cell Commun. Signal. 2019, 17, 34. [Google Scholar] [CrossRef] [Green Version]
- Saponara, S.; Sgaragli, G.; Fusi, F. Quercetin as a Novel Activator of L-Type Ca2+ Channels in Rat Tail Artery Smooth Muscle Cells: Quercetin Increases Vascular Smooth Muscle ICa(L). Br. J. Pharmacol. 2002, 135, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.T.; Bhogal, N.K.; Diakonov, I.; Pannell, L.M.K.; Perera, R.K.; Bork, N.I.; Schobesberger, S.; Lucarelli, C.; Faggian, G.; Alvarez-Laviada, A.; et al. Cardiomyocyte Membrane Structure and CAMP Compartmentation Produce Anatomical Variation in Β2AR-CAMP Responsiveness in Murine Hearts. Cell Rep. 2018, 23, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balijepalli, R.C.; Foell, J.D.; Hall, D.D.; Hell, J.W.; Kamp, T.J. Localization of Cardiac L-Type Ca2+ Channels to a Caveolar Macromolecular Signaling Complex Is Required for Beta(2)-Adrenergic Regulation. Proc. Natl. Acad. Sci. USA 2006, 103, 7500–7505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makkar, R.R. Female Gender as a Risk Factor for Torsades de Pointes Associated with Cardiovascular Drugs. J. Am. Med. Assoc. 1993, 270, 2590–2597. [Google Scholar] [CrossRef]
- Yang, P.-C.; Kurokawa, J.; Furukawa, T.; Clancy, C.E. Acute Effects of Sex Steroid Hormones on Susceptibility to Cardiac Arrhythmias: A Simulation Study. PLoS Comput. Biol. 2010, 6, e1000658. [Google Scholar] [CrossRef]
- Costa, S.; Saguner, A.M.; Gasperetti, A.; Akdis, D.; Brunckhorst, C.; Duru, F. The Link Between Sex Hormones and Susceptibility to Cardiac Arrhythmias: From Molecular Basis to Clinical Implications. Front. Cardiovasc. Med. 2021, 8, 644279. [Google Scholar] [CrossRef]
- Reineri, S.; Bertoni, A.; Sanna, E.; Baldassarri, S.; Sarasso, C.; Zanfa, M.; Canobbio, I.; Torti, M.; Sinigaglia, F. Membrane Lipid Rafts Coordinate Estrogen-Dependent Signaling in Human Platelets. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 273–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balijepalli, R.C.; Kamp, T.J. Caveolae, Ion Channels and Cardiac Arrhythmias. Prog. Biophys. Mol. Biol. 2008, 98, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Mermelstein, P.G.; Becker, J.B.; Surmeier, D.J. Estradiol Reduces Calcium Currents in Rat Neostriatal Neurons via a Membrane Receptor. J. Neurosci. 1996, 16, 595–604. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.Y.; Chai, Y.G.; Lee, E.B.; Kim, K.W.; Nah, S.-Y.; Oh, T.H.; Rhim, H. 17Beta-Estradiol Inhibits High-Voltage-Activated Calcium Channel Currents in Rat Sensory Neurons via a Non-Genomic Mechanism. Life Sci. 2002, 70, 2047–2059. [Google Scholar] [CrossRef]
- Brewer, L.D.; Dowling, A.L.S.; Curran-Rauhut, M.A.; Landfield, P.W.; Porter, N.M.; Blalock, E.M. Estradiol Reverses a Calcium-Related Biomarker of Brain Aging in Female Rats. J. Neurosci. 2009, 29, 6058–6067. [Google Scholar] [CrossRef] [Green Version]
- Kurata, K.; Takebayashi, M.; Kagaya, A.; Morinobu, S.; Yamawaki, S. Effect of β-Estradiol on Voltage-Gated Ca2+ Channels in Rat Hippocampal Neurons: A Comparison with Dehydroepiandrosterone. Eur. J. Pharmacol. 2001, 416, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Hur, E.-M.; Park, T.-J.; Kim, K.-T. Nongenomic Inhibition of Catecholamine Secretion by 17β-Estradiol in PC12 Cells. J. Neurochem. 2002, 74, 2490–2496. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ye, Q.; Lu, R.; Cao, J.; Wang, J.; Ding, H.; Gao, R.; Xiao, H. Effects of Estradiol on High-Voltage-Activated Ca2+ Channels in Cultured Rat Cortical Neurons. Endocr. Res. 2014, 39, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, J.C.; López-Zapata, D.F.; Pinzón, O.A. Effects of 17beta-Estradiol and IGF-1 on L-Type Voltage-Activated and Stretch-Activated Calcium Currents in Cultured Rat Cortical Neurons. Neuroendocrinol. Lett. 2014, 35, 724–732. [Google Scholar]
- Deutschmann, A.; Hans, M.; Meyer, R.; Häberlein, H.; Swandulla, D. Bisphenol A Inhibits Voltage-Activated Ca2+ Channels in Vitro: Mechanisms and Structural Requirements. Mol. Pharmacol. 2013, 83, 501–511. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Zhu, T.; Guo, F.; Huang, S.; Hu, H.; Feng, R.; Hao, L. Nonylphenol, an Environmental Estrogen, Affects Voltage-Gated K+ Currents and L-Type Ca2+ Currents in a Non-Monotonic Manner in GH3 Pituitary Cells. Toxicol. Lett. 2013, 218, 137–143. [Google Scholar] [CrossRef]
- Dingemans, M.M.L.; van den Berg, M.; Bergman, A.; Westerink, R.H.S. Calcium-Related Processes Involved in the Inhibition of Depolarization-Evoked Calcium Increase by Hydroxylated PBDEs in PC12 Cells. Toxicol. Sci. 2010, 114, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Langeveld, W.T.; Meijer, M.; Westerink, R.H.S. Differential Effects of 20 Non-Dioxin-Like PCBs on Basal and Depolarization-Evoked Intracellular Calcium Levels in PC12 Cells. Toxicol. Sci. 2012, 126, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.N.; Huang, R.-Q.; Logan, S.M.; Yi, K.D.; Dillon, G.H.; Simpkins, J.W. Estrogens Directly Potentiate Neuronal L-Type Ca2+ Channels. Proc. Natl. Acad. Sci. USA 2008, 105, 15148–15153. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Chu, Z.; Moenter, S.M. Diurnal In Vivo and Rapid In Vitro Effects of Estradiol on Voltage-Gated Calcium Channels in Gonadotropin-Releasing Hormone Neurons. J. Neurosci. 2010, 30, 3912–3923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, I.; Sárvári, M.; Aller, M.; Okada, N.; Okada, H.; Likó, I.; Liposits, Z. Estrogen Receptor α and β Differentially Mediate C5aR Agonist Evoked Ca2+-Influx in Neurons through L-Type Voltage-Gated Ca2+ Channels. Neurochem. Int. 2012, 60, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Sedej, S.; Tsujimoto, T.; Zorec, R.; Rupnik, M. Voltage-Activated Ca2+ Channels and Their Role in the Endocrine Function of the Pituitary Gland in Newborn and Adult Mice: VACCs in the Endocrine Function of the Mouse Pituitary Gland. J. Physiol. 2004, 555, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Bosch, M.A.; Jamali, K.; Xue, C.; Kelly, M.J.; Ronnekleiv, O.K. Estrogen Upregulates T-Type Calcium Channels in the Hypothalamus and Pituitary. J. Neurosci. 2006, 26, 11072–11082. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.A.; Mamounis, K.J.; Yasrebi, A.; Roepke, T.A. Regulation of Gene Expression by 17β-Estradiol in the Arcuate Nucleus of the Mouse through ERE-Dependent and ERE-Independent Mechanisms. Steroids 2016, 107, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.W.; Kyrozis, A.; Chevaleyre, V.; Kow, L.-M.; Zhou, J.; Devidze, N.; Zhang, Q.; Etgen, A.M.; Pfaff, D.W. Voltage-Dependent Calcium Channels in Ventromedial Hypothalamic Neurones of Postnatal Rats: Modulation by Oestradiol and Phenylephrine. J. Neuroendocrinol. 2008, 20, 188–198. [Google Scholar] [CrossRef]
- Zhang, C.; Bosch, M.A.; Rick, E.A.; Kelly, M.J.; Rønnekleiv, O.K. 17β-Estradiol Regulation of T-Type Calcium Channels in Gonadotropin-Releasing Hormone Neurons. J. Neurosci. 2009, 29, 10552–10562. [Google Scholar] [CrossRef] [Green Version]
- Bosch, M.A.; Hou, J.; Fang, Y.; Kelly, M.J.; RØnnekleiv, O.K. 17β-Estradiol Regulation of the MRNA Expression of t-Type Calcium Channel Subunits: Role of Estrogen Receptor α and Estrogen Receptor β. J. Comp. Neurol. 2009, 512, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Gieske, M.C.; Trudgen, K.L.; Hudgins-Spivey, S.; Kim, B.G.; Krust, A.; Chambon, P.; Jeong, J.-W.; Blalock, E.; Ko, C. Identification of Estradiol/ERα-Regulated Genes in the Mouse Pituitary. J. Endocrinol. 2011, 210, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, B.; Du, F.; Li, H.; Wang, S.; Hu, C.; Zhu, C.; Yu, X. The Involvement of PI3K-Mediated and L-VGCC-Gated Transient Ca2+ Influx in 17β-Estradiol-Mediated Protection of Retinal Cells from H2O2-Induced Apoptosis with Ca2+ Overload. PLoS ONE 2013, 8, e77218. [Google Scholar] [CrossRef] [Green Version]
- Ruffinatti, F.A.; Gilardino, A.; Secchi, V.; Cottone, E.; Lovisolo, D.; Bovolin, P. Bisphenol A Activates Calcium Influx in Immortalized GnRH Neurons. Int. J. Mol. Sci. 2019, 20, 2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnoult, C.; Cardullo, R.A.; Lemos, J.R.; Florman, H.M. Activation of Mouse Sperm T-Type Ca2+ Channels by Adhesion to the Egg Zona Pellucida. Proc. Natl. Acad. Sci. USA 1996, 93, 13004–13009. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, F.; López-González, I.; Muñoz-Garay, C.; Felix, R.; De la Vega-Beltrán, J.L.; Kopf, G.S.; Visconti, P.E.; Darszon, A. Dual Regulation of the T-Type Ca 2+ Current by Serum Albumin and β-Estradiol in Mammalian Spermatogenic Cells. FEBS Lett. 2000, 475, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Lu, L.; Gao, X.; Wang, C.; Wang, J.; Cheng, J.; Gao, R.; Xiao, H. Effects of Raloxifene on Voltage- Dependent T-Type Ca2+ Channels in Mouse Spermatogenic Cells. Pharmacology 2011, 87, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Banciu, A.; Banciu, D.D.; Mustaciosu, C.C.; Radu, M.; Cretoiu, D.; Xiao, J.; Cretoiu, S.M.; Suciu, N.; Radu, B.M. Beta-Estradiol Regulates Voltage-Gated Calcium Channels and Estrogen Receptors in Telocytes from Human Myometrium. Int. J. Mol. Sci. 2018, 19, 1413. [Google Scholar] [CrossRef] [Green Version]
- Klein, M.; Csöbönyeiová, M.; Danišovič, Ľ.; Lapides, L.; Varga, I. Telocytes in the Female Reproductive System: Up-to-Date Knowledge, Challenges and Possible Clinical Applications. Life 2022, 12, 267. [Google Scholar] [CrossRef]
- Azenabor, A.A.; Chaudhry, A.U. 17β-Estradiol Induces L-Type Ca2+ Channel Activation and Regulates Redox Function in Macrophages. J. Reprod. Immunol. 2003, 59, 17–28. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, Y.; Xie, K.; Sun, X.; Gao, Y.; Wang, Z. Estrogen-Induced Nongenomic Calcium Signaling Inhibits Lipopolysaccharide-Stimulated Tumor Necrosis Factor α Production in Macrophages. PLoS ONE 2013, 8, e83072. [Google Scholar] [CrossRef] [Green Version]
- Ruehlmann, D.O.; Steinert, J.R.; Valverde, M.A.; Jacob, R.; Mann, G.E. Environmental Estrogenic Pollutants Induce Acute Vascular Relaxation by Inhibiting L-type Ca2+ Channels in Smooth Muscle Cells. FASEB J. 1998, 12, 613–619. [Google Scholar] [CrossRef]
- Gao, Q.; Liu, S.; Guo, F.; Liu, S.; Yu, X.; Hu, H.; Sun, X.; Hao, L.; Zhu, T. Nonylphenol Affects Myocardial Contractility and L-Type Ca2+ Channel Currents in a Non-Monotonic Manner via G Protein-Coupled Receptor 30. Toxicology 2015, 334, 122–129. [Google Scholar] [CrossRef]
- Liang, Q.; Gao, X.; Chen, Y.; Hong, K.; Wang, H.-S. Cellular Mechanism of the Nonmonotonic Dose Response of Bisphenol A in Rat Cardiac Myocytes. Environ. Health Perspect. 2014, 122, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Araújo, I.G.A.; Silva, D.F.; do Carmo de Alustau, M.; Dias, K.L.G.; Cavalcante, K.V.M.; Veras, R.C.; Barbosa-Filho, J.M.; Neto, M.D.A.; Bendhack, L.M.; de Azevedo Correia, N.; et al. Calcium Influx Inhibition Is Involved in the Hypotensive and Vasorelaxant Effects Induced by Yangambin. Molecules 2014, 19, 6863–6876. [Google Scholar] [CrossRef] [PubMed]
- Schrank, S.; Barrington, N.; Stutzmann, G.E. Calcium-Handling Defects and Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2020, 12, a035212. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lu, M.; Lancaster, T.; Cao, P.; Honda, S.-I.; Staufenbiel, M.; Harada, N.; Zhong, Z.; Shen, Y.; Li, R. Brain Estrogen Deficiency Accelerates Aβ Plaque Formation in an Alzheimer’s Disease Animal Model. Proc. Natl. Acad. Sci. USA 2005, 102, 19198–19203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.C.; Rosario, E.R.; Chang, L.; Stanczyk, F.Z.; Oddo, S.; LaFerla, F.M.; Pike, C.J. Progesterone and Estrogen Regulate Alzheimer-Like Neuropathology in Female 3xTg-AD Mice. J. Neurosci. 2007, 27, 13357–13365. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.-J.; Zhu, B.-L.; Sun, F.; Luo, D.; Ma, Y.-L.; Luo, B.; Tang, J.; Xiong, M.-J.; Liu, L.; Long, Y.; et al. Estrogen Receptor α Promotes Cav1.2 Ubiquitination and Degradation in Neuronal Cells and in APP/PS1 Mice. Aging Cell 2019, 18, e12961. [Google Scholar] [CrossRef]
- Sribnick, E.A.; Del Re, A.M.; Ray, S.K.; Woodward, J.J.; Banik, N.L. Estrogen Attenuates Glutamate-Induced Cell Death by Inhibiting Ca2+ Influx through L-Type Voltage-Gated Ca2+ Channels. Brain Res. 2009, 1276, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Russo, J.; Russo, I.H. The Role of Estrogen in the Initiation of Breast Cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 89–96. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.B.; Hankinson, S.E. Endogenous Estrogens and the Risk of Breast, Endometrial, and Ovarian Cancers. Steroids 2015, 99, 8–10. [Google Scholar] [CrossRef]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen Signaling in Endometrial Cancer: A Key Oncogenic Pathway with Several Open Questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Tajada, S.; Villalobos, C. Calcium Permeable Channels in Cancer Hallmarks. Front. Pharmacol. 2020, 11, 968. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.J.; Hernández-Ochoa, E.; Martin, S.S. Calcium Signaling: Breast Cancer’s Approach to Manipulation of Cellular Circuitry. Biophys. Rev. 2020, 12, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Bao, X.; Jin, B.; Wang, X.; Mao, Z.; Li, X.; Wei, L.; Shen, D.; Wang, J. Ca2+ Channel Subunit α 1D Promotes Proliferation and Migration of Endometrial Cancer Cells Mediated by 17β-estradiol via the G Protein-coupled Estrogen Receptor. FASEB J. 2015, 29, 2883–2893. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Han, Z.; Shao, L.; Zhao, Y. Ultrasound-Targeted Microbubble Destruction of Calcium Channel Subunitα 1D SiRNA Inhibits Breast Cancer via GProtein-Coupled Receptor 30. Oncol. Rep. 2016, 36, 1886–1892. [Google Scholar] [CrossRef] [Green Version]
- Pera, E. The Voltage Gated Ca2+-Channel Cav3.2 and Therapeutic Responses in Breast Cancer. Cancer Cell Int. 2016, 16, 24. [Google Scholar] [CrossRef] [Green Version]
- Sekar, S.; Subbamanda, Y.; Pullaguri, N.; Sharma, A.; Sahu, C.; Kumar, R.; Bhargava, A. Isoform-Specific Expression of T-Type Voltage-Gated Calcium Channels and Estrogen Receptors in Breast Cancer Reveals Specific Isoforms That May Be Potential Targets. Curr. Res. Biotechnol. 2022, 4, 459–467. [Google Scholar] [CrossRef]
- Filardo, E.J.; Graeber, C.T.; Quinn, J.A.; Resnick, M.B.; Giri, D.; DeLellis, R.A.; Steinhoff, M.M.; Sabo, E. Distribution of GPR30, a Seven Membrane-Spanning Estrogen Receptor, in Primary Breast Cancer and Its Association with Clinicopathologic Determinants of Tumor Progression. Clin. Cancer Res. 2006, 12, 6359–6366. [Google Scholar] [CrossRef] [Green Version]
- Marjon, N.A.; Hu, C.; Hathaway, H.J.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor Regulates Mammary Tumorigenesis and Metastasis. Mol. Cancer Res. 2014, 12, 1644–1654. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Ma, D.; Chen, S.; Tang, R.; Yang, J.; Meng, C.; Feng, Y.; Liu, L.; Wang, J.; Luo, H.; et al. High GPER Expression in Triple-Negative Breast Cancer Is Linked to pro-Metastatic Pathways and Predicts Poor Patient Outcomes. NPJ Breast Cancer 2022, 8, 100. [Google Scholar] [CrossRef]
- Jakab, M.; Lach, S.; Bacová, Z.; Langelüddecke, C.; Strbák, V.; Schmidt, S.; Iglseder, E.; Paulmichl, M.; Geibel, J.; Ritter, M. Resveratrol Inhibits Electrical Activity and Insulin Release from Insulinoma Cells by Block of Voltage-Gated Ca2+ Channels and Swelling-Dependent Cl- Currents. Cell Physiol. Biochem. 2008, 22, 567–578. [Google Scholar] [CrossRef]
- Bardy, G.; Virsolvy, A.; Quignard, J.F.; Ravier, M.A.; Bertrand, G.; Dalle, S.; Cros, G.; Magous, R.; Richard, S.; Oiry, C. Quercetin Induces Insulin Secretion by Direct Activation of L-Type Calcium Channels in Pancreatic Beta Cells: Quercetin Increases L-Type Ca Currents in Beta Cells. Br. J. Pharmacol. 2013, 169, 1102–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C.; Nagpal, M.L.; Stocco, D.M.; Lin, T. Effects of Genistein, Resveratrol, and Quercetin on Steroidogenesis and Proliferation of MA-10 Mouse Leydig Tumor Cells. J. Endocrinol. 2007, 192, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhang, Y.; Li, S.; Sun, W.; Soong, T.W. Tyrosine Kinase-Independent Inhibition by Genistein on Spermatogenic T-Type Calcium Channels Attenuates Mouse Sperm Motility and Acrosome Reaction. Cell Calcium 2009, 45, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Schiff, R. Growth Factor Receptor Cross-Talk with Estrogen Receptor as a Mechanism for Tamoxifen Resistance in Breast Cancer. Breast 2003, 12, 362–367. [Google Scholar] [CrossRef]
- Ruan, W.; Catanese, V.; Wieczorek, R.; Feldman, M.; Kleinberg, D.L. Estradiol Enhances the Stimulatory Effect of Insulin-like Growth Factor-I (IGF-I) on Mammary Development and Growth Hormone-Induced IGF-I Messenger Ribonucleic Acid. Endocrinology 1995, 136, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the Estrogen Receptor and the HER Tyrosine Kinase Receptor Family: Molecular Mechanism and Clinical Implications for Endocrine Therapy Resistance. Endocr. Rev. 2008, 29, 217–233. [Google Scholar] [CrossRef] [Green Version]
- Giuliano, M.; Trivedi, M.V.; Schiff, R. Bidirectional Crosstalk between the Estrogen Receptor and Human Epidermal Growth Factor Receptor 2 Signaling Pathways in Breast Cancer: Molecular Basis and Clinical Implications. Breast Care 2013, 8, 256–262. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, A.; Lin, X.; Novak, P.; Mehta, K.; Korchev, Y.; Delmar, M.; Gorelik, J. Super-Resolution Scanning Patch Clamp Reveals Clustering of Functional Ion Channels in Adult Ventricular Myocyte. Circ. Res. 2013, 112, 1112–1120. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Channel | Current Type | Localization |
|---|---|---|
| CaV1.1 | L | Skeletal muscle [11] |
| CaV1.2 | L | Heart, smooth muscle, brain, pituitary, pancreatic β-cells, adrenal medulla [8,19,20] |
| CaV1.3 | L | Brain, pancreatic β-cells, medulla, kidney, ovary, cochlea [8,21,22] |
| CaV1.4 | L | Retina [10] |
| CaV2.1 | P/Q | Central nervous system, cerebellum, cochlea, adrenal medulla [16,23,24] |
| CaV2.2 | N | Brain, peripheral nervous system, adrenal medulla [23] |
| CaV2.3 | R | Central nervous system, pancreatic islets, adrenal medulla [18,23,24] |
| CaV3.1 | T | Brain, ovary, placenta, heart, peripheral nervous system, pancreatic β cells, adrenal medulla [25,26] |
| CaV3.2 | T | Heart, brain, kidney, pancreatic β cells, adrenal cortex [27,28] |
| CaV3.3 | T | Brain, pancreatic β cells [29,30] |
| Effect of Estrogen on VGCCs | Experimental System | Mechanism of Action | Remarks |
|---|---|---|---|
| VGCC inhibition | Rabbit coronary [112] and basilar artery [113], vascular smooth muscle cells from Sprague Dawley rats [106], guinea pig ventricular myocytes [107] | Non-genomic/(not-specified) | Estrogen inhibited the calcium influx via VGCCs and thereby caused vasodilation. This inhibitory effect was reported to occur via a pertussis toxin-sensitive GTP-binding protein [113]. |
| LTCC inhibition | Rat aortic smooth muscle (A7r5 cells) [111], mammalian smooth muscles [114], ventricular myocytes from rat, human and guinea pig [109,110], rabbit carotid artery [115], neonatal rat cardiac fibroblasts [108], HEK-293 cells transiently transfected with human Cav1.2α [121] | Non-genomic/receptor-independent | Estrogen reduced calcium influx through inhibition of LTCCs in various cardiovascular tissues. |
| Right coronary artery from female Yorkshire pigs [126] | ER-dependent | Estrogen reduced the LTCC protein expression via ERα/ERβ dependent pathway. Estrogen binds to ERα/ERβ and alters the post-transcriptional regulation of LTCC. | |
| TTCC inhibition | Neonatal cardiomyocytes from female Wistar rats [124] | Receptor-independent | TTCC downregulation by estrogen was mediated by ERK-1/2, 5 pathways. |
| LTCC and TTCC inhibition | A7r5 vascular smooth muscle cell line [120] | Not specified | Estrogen application attenuated the voltage-dependent calcium current (within 1–2 min) through TTCCs and LTCCs. |
| LTCC upregulation | Ventricular myocytes from adult male and female New Zealand white rabbits [127,128,129] and human iPSC derived cardiac myocytes. | ER-dependent | Physiological concentration of estrogen (1 nM) increased the calcium current only in cells from the base of the heart. This estrogenic effect could be correlated to humans [129]. |
| Rat ventricular myocytes, H9C2 cultured cells [130] | Membrane ER-dependent | Upregulation of calcium influx via LTCCs occurred via plasma membrane ER and by activation of PI3K, protein kinase B (Akt/PKB) and cAMP- response element binding protein (CREB) signaling. | |
| Mice left ventricular apical myocytes [131] | Non-genomic (GPR30) | Estrogen modulated the expression of genes related to the cAMP-PKA-LTCC pathway thereby contributing to sex differences in cardiac contraction. This acute estrogenic effect was concentration-dependent, sex-specific and mediated by GPR30. |
| Effect of Estrogen on VGCCs | Experimental System | Mechanism of Action | Remarks |
|---|---|---|---|
| HVA VGCC inhibition | Female rat cortical neurons [145] | Non-genomic | Estrogen inhibited HVA calcium current in a rapid, reversible and concentration-dependent manner via PKC and PKA-dependent pathways. |
| N-and LTCC inhibition | Sensory neurons of female Sprague Dawley rats [141] | Non-genomic | First evidence of linking modulation of HVA L-and N-type calcium currents by estrogen to in vivo sensory modulation. |
| LTCC inhibition | Hippocampal zipper slices from female Fischer rats [142] | Not specified | Estrogen inhibited LTCC CaV1.2, but not CaV1.3. |
| Neostriatal neurons from Sprague Dawley rats [140], hippocampal cells from Wistar rats [143], neuronal cells from Wistar rat cortex [146] | Non-genomic/membrane receptor mediated | Estrogen inhibited LTCCs via a non-genomic mechanism. ER antagonists or inhibitors of PKA/PKC did not affect the estrogen-mediated inhibition of HVA LTCCs, suggesting that more than one mechanism may be operating in neuronal tissues. | |
| GnRH producing GT-17 neuronal cells [153] | ER-dependent | Estrogen reduced the transcription of CaV1.3 LTCC. | |
| LTCC upregulation | Rat hippocampal neurons, hippocampal slices, and HEK-293 cells transfected with neuronal LTCCs [151] | ER-independent/membrane receptor mediated | Estrogen directly potentiated recombinant CaV1.2 in the hippocampal neurons via an ER-independent mechanism through direct binding with a domain that overlaps the dihydropyridine-binding site [151]. |
| L-and R-type VGCC upregulation | GnRH neurons from adult female mice expressing eGFP [152] | ERβ and GPR30 | Estrogen rapidly increased the inward calcium currents through L-and R-type channels by activating ERβ and GPR30, respectively. |
| TTCC upregulation | Adult C57BL/6 mice hypothalamic arcuate nucleus [156], mice hypothalamic nuclei and pituitary [159] | ER-dependent | Estrogen-induced increase in mRNA expression of CaV3.1 and CaV3.2 in the hypothalamus was dependent on ERα and both (ERα and ERβ), respectively. However, in the pituitary, the estrogenic effect was dependent on the expression of ERα alone [159]. |
| Mice-GnRH neurons [158] | Membrane ER | All three TTCC isoforms are expressed in GnRH neurons and the estrogen-dependent upregulation of TTCC is membrane ER-mediated. | |
| Sprague Dawley rat ventromedial hypothalamic neurons [157], Guinea pig hypothalamus and pituitary neurons [155] | Not specified | Estrogen enhanced LVA calcium current in the absence of phenylephrine, an α1adrenergic agonist. In contrast, in its presence, augmentation of HVA calcium currents mediated by N-and L-type VGCCs was observed [157]. | |
| P/Q and TTCC upregulation | C57BL/6 mice pituitary [160] | ERα-dependent | Estrogen regulated P/Q and TTCCs via ERα-mediated pathway in the pituitary. |
| In Vitro Models or Methods | |
|---|---|
| Experimental System/Tools | Potential Effects That Can Be Studied |
| Heterologous overexpression of ion channels in cell lines such as HEK293 and COS-7, primary cell lines. |
|
| Native cell lines (e.g., INS-1E, PC12). |
|
| In Vivo/Ex Vivo Models or Methods | |
| Transgenic ligand deficient models (e.g., ovariectomized (ER-deficient animal model), disease models (e.g., models for Parkinson’s or Alzheimer’s). Brain slices for patch clamp recording of intact (DRG) neurons. Whole organ systems such as Langendorff heart. |
|
| Alternate Models or Methods | |
| Economic zebrafish models |
|
| Super-resolution scanning patch-clamp | Microdomain-dependent estrogen regulation of ion channels. |
| Bio-layer interferometry (BLI) | Biomolecular interactions for investigating the binding sites of estrogen/ligand on ion channels in cell lysates. |
| Functional in-silico models (such as the recent A549 in-silico whole-cell ion current model) | The alterations in ion channels caused by various stimuli can be investigated with a digital model prior to experimental validation in the native background. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbamanda, Y.D.; Bhargava, A. Intercommunication between Voltage-Gated Calcium Channels and Estrogen Receptor/Estrogen Signaling: Insights into Physiological and Pathological Conditions. Cells 2022, 11, 3850. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233850
Subbamanda YD, Bhargava A. Intercommunication between Voltage-Gated Calcium Channels and Estrogen Receptor/Estrogen Signaling: Insights into Physiological and Pathological Conditions. Cells. 2022; 11(23):3850. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233850
Chicago/Turabian StyleSubbamanda, Yashashwini Dinesh, and Anamika Bhargava. 2022. "Intercommunication between Voltage-Gated Calcium Channels and Estrogen Receptor/Estrogen Signaling: Insights into Physiological and Pathological Conditions" Cells 11, no. 23: 3850. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233850