
Protein Kinase D1 Signaling in Cancer Stem Cells with Epithelial-Mesenchymal Plasticity

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Key Reagents and Antibodies
2.2. Cell Culture
2.3. Real Time RT-qPCR
2.4. Immunoblot Assays
2.5. Human pNET Specimens
2.6. Immunofluorescence and Immunohistochemistry
2.7. Aldehyde Dehydrogenase (ALDH) Activity Assays
2.8. Tumorsphere Formation Assays
2.9. In Vitro Extreme Limiting Dilution and Tumorsphere Formation Assays
2.10. Statistics
3. Results
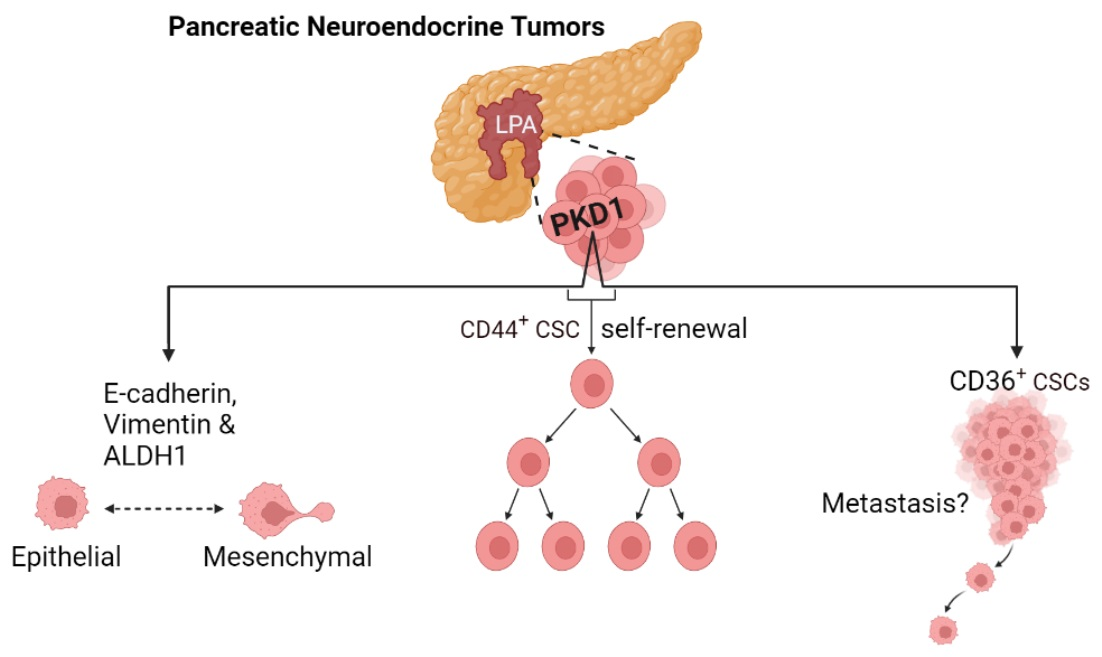
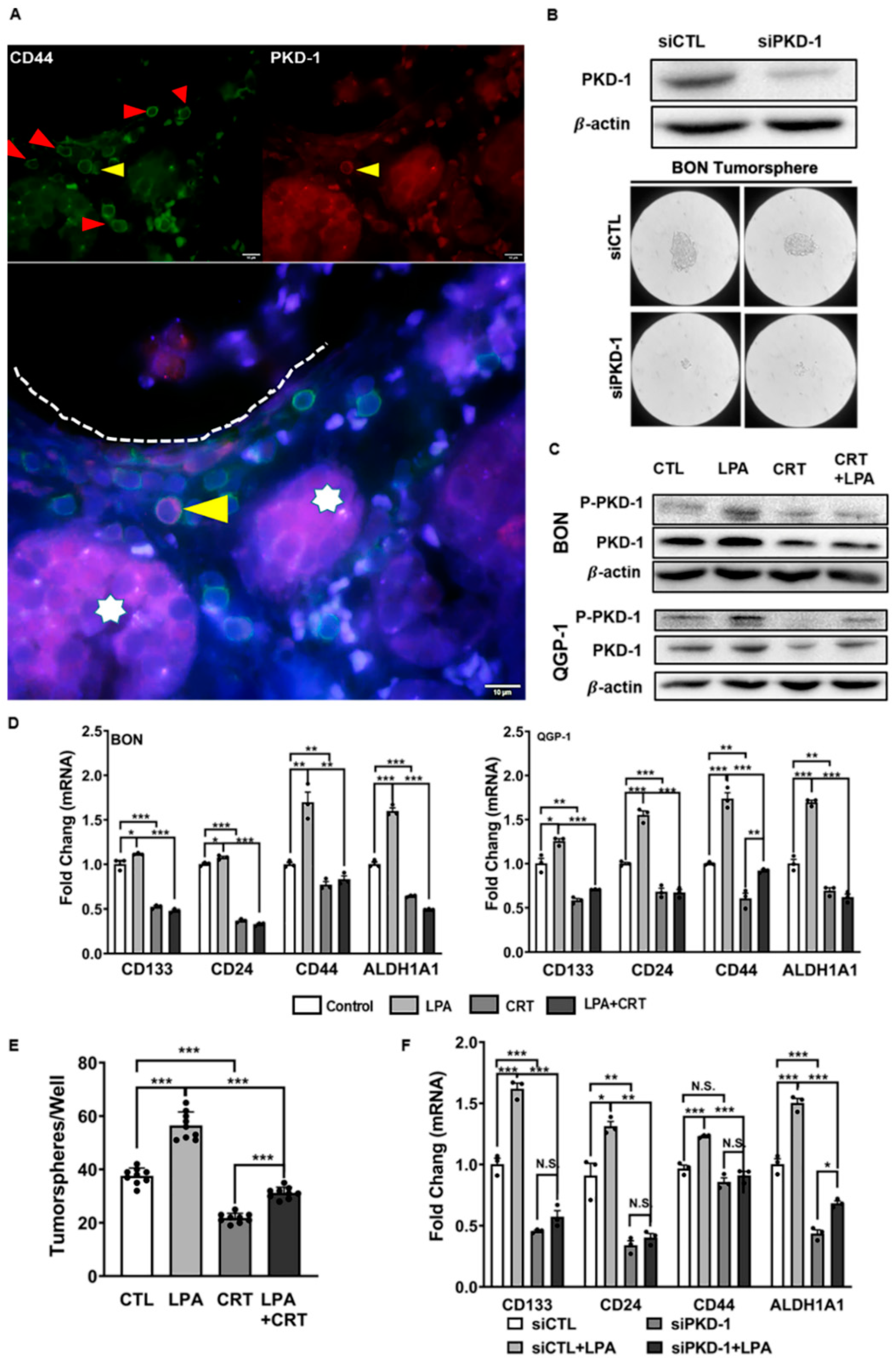
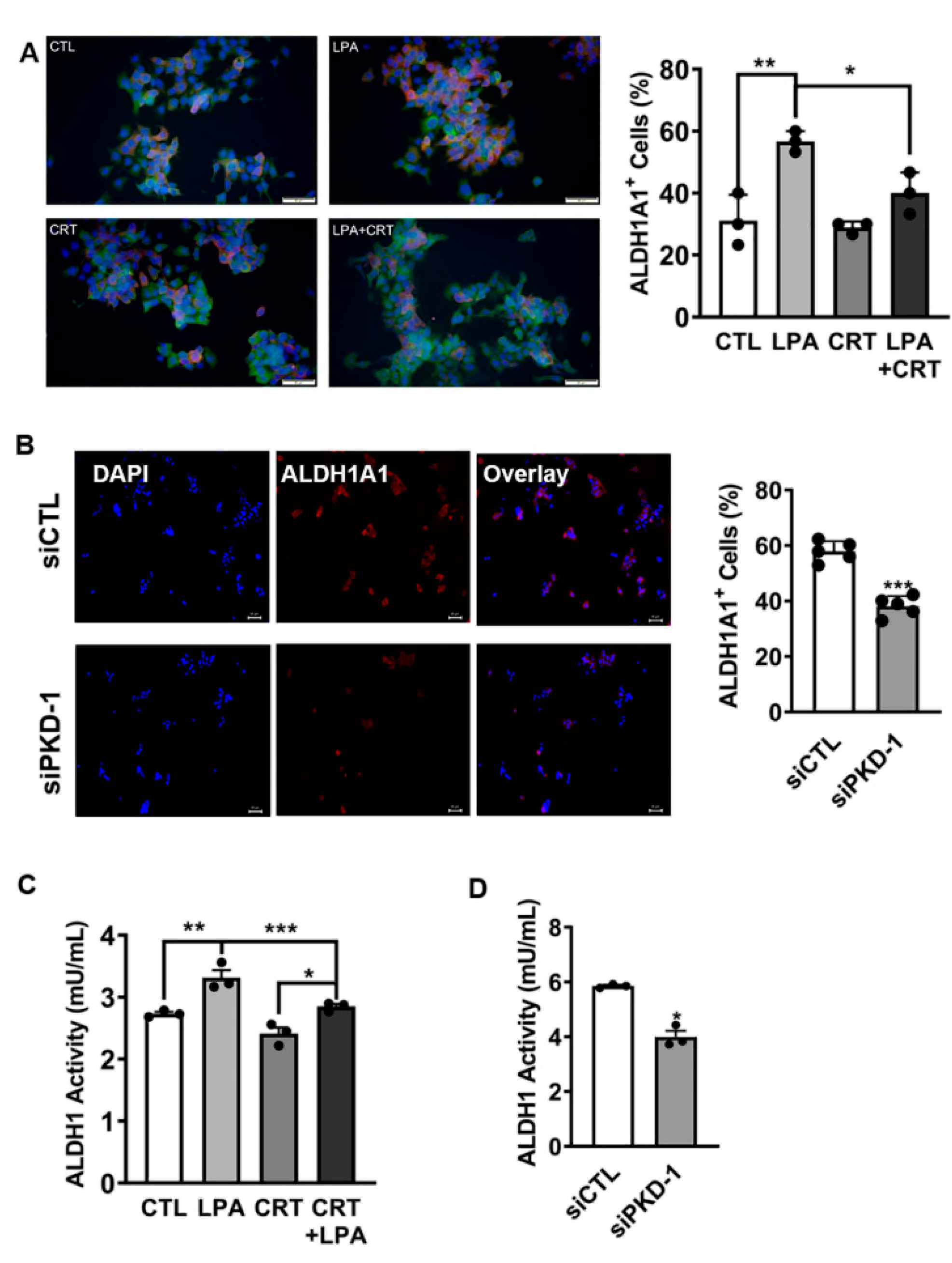
3.1. Regulation of Stem-like Phenotype in pNETs by PKD1 Signaling
3.2. Requirement of PKD1 Signaling in Partial EMT and CSC Plasticity
3.3. Critical Role of PKD1 Signaling in CD36 Expression in pNET Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Folkman, J.; Watson, K.; Ingber, D.; Hanahan, D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Sadanandam, A.; Wullschleger, S.; Lyssiotis, C.A.; Grotzinger, C.; Barbi, S.; Bersani, S.; Korner, J.; Wafy, I.; Mafficini, A.; Lawlor, R.T.; et al. A Cross-Species Analysis in Pancreatic Neuroendocrine Tumors Reveals Molecular Subtypes with Distinctive Clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov. 2015, 5, 1296–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Rose, J.B.; Liu, Y.; Jaskular-Sztul, R.; Contreras, C.; Beck, A.; Chen, H. Heterogeneity of Vascular Endothelial Cells, De Novo Arteriogenesis and Therapeutic Implications in Pancreatic Neuroendocrine Tumors. J. Clin. Med. 2019, 8, 1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharjan, C.K.; Ear, P.H.; Tran, C.G.; Howe, J.R.; Chandrasekharan, C.; Quelle, D.E. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers 2021, 13, 5117. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Terris, B.; Scoazec, J.Y.; Rubbia, L.; Bregeaud, L.; Pepper, M.S.; Ruszniewski, P.; Belghiti, J.; Flejou, J.; Degott, C. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology 1998, 32, 133–138. [Google Scholar] [CrossRef]
- Ren, B. Protein Kinase D1 Signaling in Angiogenic Gene Expression and VEGF-Mediated Angiogenesis. Front. Cell Dev. Biol. 2016, 4, 37. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Capozzi, M.; Von Arx, C.; De Divitiis, C.; Ottaiano, A.; Tatangelo, F.; Romano, G.M.; Tafuto, S. Antiangiogenic Therapy in Pancreatic Neuroendocrine Tumors. Anticancer Res. 2016, 36, 5025–5030. [Google Scholar] [CrossRef]
- Cho, C.M. Recent Updates in the Management of Advanced Pancreatic Neuroendocrine Tumors. Korean J. Gastroenterol. 2019, 73, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Buicko, J.L.; Finnerty, B.M.; Zhang, T.; Kim, B.J.; Fahey, T.J., 3rd; Nancy Du, Y.C. Insights into the biology and treatment strategies of pancreatic neuroendocrine tumors. Ann. Pancreat. Cancer 2019, 2, 12. [Google Scholar] [CrossRef]
- Li, F.; Xu, J.; Liu, S. Cancer Stem Cells and Neovascularization. Cells 2021, 10, 1070. [Google Scholar] [CrossRef]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Cao, Z.; Scandura, J.M.; Inghirami, G.G.; Shido, K.; Ding, B.S.; Rafii, S. Molecular Checkpoint Decisions Made by Subverted Vascular Niche Transform Indolent Tumor Cells into Chemoresistant Cancer Stem Cells. Cancer Cell 2017, 31, 110–126. [Google Scholar] [CrossRef] [Green Version]
- Akil, A.; Gutierrez-Garcia, A.K.; Guenter, R.; Rose, J.B.; Beck, A.W.; Chen, H.; Ren, B. Notch Signaling in Vascular Endothelial Cells, Angiogenesis, and Tumor Progression: An Update and Prospective. Front. Cell Dev. Biol. 2021, 9, 642352. [Google Scholar] [CrossRef]
- Jiang, Y.; Guo, Y.; Hao, J.; Guenter, R.; Lathia, J.; Beck, A.W.; Hattaway, R.; Hurst, D.; Wang, Q.J.; Liu, Y.; et al. Development of an arteriolar niche and self-renewal of breast cancer stem cells by lysophosphatidic acid/protein kinase D signaling. Commun. Biol. 2021, 4, 780. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Gaur, P.; Sceusi, E.L.; Samuel, S.; Xia, L.; Fan, F.; Zhou, Y.; Lu, J.; Tozzi, F.; Lopez-Berestein, G.; Vivas-Mejia, P.; et al. Identification of cancer stem cells in human gastrointestinal carcinoid and neuroendocrine tumors. Gastroenterology 2011, 141, 1728–1737. [Google Scholar] [CrossRef]
- Krampitz, G.W.; George, B.M.; Willingham, S.B.; Volkmer, J.P.; Weiskopf, K.; Jahchan, N.; Newman, A.M.; Sahoo, D.; Zemek, A.J.; Yanovsky, R.L.; et al. Identification of tumorigenic cells and therapeutic targets in pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 4464–4469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mafficini, A.; Scarpa, A. Genomic landscape of pancreatic neuroendocrine tumours: The International Cancer Genome Consortium. J. Endocrinol. 2018, 236, R161–R167. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.A.; Liu, P.; Spitaler, M.; Olson, E.N.; McKinsey, T.A.; Cantrell, D.A.; Scharenberg, A.M. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol. Cell Biol. 2006, 26, 1569–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Best, B.; Ramakrishnan, D.P.; Walcott, B.P.; Storz, P.; Silverstein, R.L. LPA/PKD-1-FoxO1 Signaling Axis Mediates Endothelial Cell CD36 Transcriptional Repression and Proangiogenic and Proarteriogenic Reprogramming. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Ye, J.; Deng, F.; Wang, Q.J. Protein kinase D signaling in cancer: A friend or foe? Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 283–294. [Google Scholar] [CrossRef]
- Zhang, X.; Connelly, J.; Chao, Y.; Wang, Q.J. Multifaceted Functions of Protein Kinase D in Pathological Processes and Human Diseases. Biomolecules 2021, 11, 483. [Google Scholar] [CrossRef]
- Ren, B.; Hale, J.; Srikanthan, S.; Silverstein, R.L. Lysophosphatidic acid suppresses endothelial cell CD36 expression and promotes angiogenesis via a PKD-1-dependent signaling pathway. Blood 2011, 117, 6036–6045. [Google Scholar] [CrossRef] [Green Version]
- Best, B.; Moran, P.; Ren, B. VEGF/PKD-1 signaling mediates arteriogenic gene expression and angiogenic responses in reversible human microvascular endothelial cells with extended lifespan. Mol. Cell Biochem. 2018, 446, 199–207. [Google Scholar] [CrossRef]
- Dong, L.; Yuan, Y.; Opansky, C.; Chen, Y.; Aguilera-Barrantes, I.; Wu, S.; Yuan, R.; Cao, Q.; Cheng, Y.C.; Sahoo, D.; et al. Diet-induced obesity links to ER positive breast cancer progression via LPA/PKD-1-CD36 signaling-mediated microvascular remodeling. Oncotarget 2017, 8, 22550–22562. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.M.; Jaskula-Sztul, R.; Ahmed, K.; Harrison, A.D.; Kunnimalaiyaan, M.; Chen, H. Resveratrol induces differentiation markers expression in anaplastic thyroid carcinoma via activation of Notch1 signaling and suppresses cell growth. Mol. Cancer Ther. 2013, 12, 1276–1287. [Google Scholar] [CrossRef]
- Hao, F.; Liu, Q.; Zhang, F.; Du, J.; Dumire, A.; Xu, X.; Cui, M.Z. LPA1-mediated PKD2 activation promotes LPA-induced tissue factor expression via the p38alpha and JNK2 MAPK pathways in smooth muscle cells. J. Biol. Chem. 2021, 297, 101152. [Google Scholar] [CrossRef]
- Panupinthu, N.; Lee, H.Y.; Mills, G.B. Lysophosphatidic acid production and action: Critical new players in breast cancer initiation and progression. Br. J. Cancer 2010, 102, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Tigyi, G.J.; Yue, J.; Norman, D.D.; Szabo, E.; Balogh, A.; Balazs, L.; Zhao, G.; Lee, S.C. Regulation of tumor cell—Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul. 2019, 71, 183–193. [Google Scholar] [CrossRef]
- Aiello, S.; Casiraghi, F. Lysophosphatidic Acid: Promoter of Cancer Progression and of Tumor Microenvironment Development. A Promising Target for Anticancer Therapies? Cells 2021, 10, 1390. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.H.; Suh, D.S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem. Cells 2016, 34, 551–564. [Google Scholar] [CrossRef]
- Li, J.; Chen, L.A.; Townsend, C.M., Jr.; Evers, B.M. PKD1, PKD2, and their substrate Kidins220 regulate neurotensin secretion in the BON human endocrine cell line. J. Biol. Chem. 2008, 283, 2614–2621. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Doppler, H.; Braun, U.B.; Panayiotou, R.; Scotti Buzhardt, M.; Radisky, D.C.; Crawford, H.C.; Fields, A.P.; Murray, N.R.; Wang, Q.J.; et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 2015, 6, 6200. [Google Scholar] [CrossRef] [Green Version]
- Kim do, Y.; Park, E.Y.; Chang, E.; Kang, H.G.; Koo, Y.; Lee, E.J.; Ko, J.Y.; Kong, H.K.; Chun, K.H.; Park, J.H. A novel miR-34a target, protein kinase D1, stimulates cancer stemness and drug resistance through GSK3/beta-catenin signaling in breast cancer. Oncotarget 2016, 7, 14791–14802. [Google Scholar] [CrossRef]
- Fleming Martinez, A.K.; Doppler, H.R.; Bastea, L.I.; Edenfield, B.; Patel, T.; Leitges, M.; Liou, G.Y.; Storz, P. Dysfunctional EGFR and oxidative stress-induced PKD1 signaling drive formation of DCLK1+ pancreatic stem cells. iScience 2021, 24, 102019. [Google Scholar] [CrossRef]
- Gimple, R.C.; Yang, K.; Halbert, M.E.; Agnihotri, S.; Rich, J.N. Brain cancer stem cells: Resilience through adaptive plasticity and hierarchical heterogeneity. Nat. Rev. Cancer 2022, 22, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Zheng-Pywell, R.; Lopez-Aguiar, A.; Fields, R.C.; Vickers, S.; Yates, C.; Dudeja, V.; Chen, H.; Reddy, S.; Maithel, S.K.; Rose, J.B. Are We Undertreating Black Patients with Nonfunctional Pancreatic Neuroendocrine Tumors? Critical Analysis of Current Surveillance Guidelines by Race. J. Am. Coll. Surg. 2022, 234, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.-C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Capodanno, Y.; Buishand, F.O.; Pang, L.Y.; Kirpensteijn, J.; Mol, J.A.; Argyle, D.J. Notch pathway inhibition targets chemoresistant insulinoma cancer stem cells. Endocr. Relat. Cancer 2018, 25, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Sharife, H.; Kreisel, T.; Mogilevsky, M.; Bar-Lev, L.; Grunewald, M.; Aizenshtein, E.; Karni, R.; Paldor, I.; Shlomi, T.; et al. Intra-Tumoral Metabolic Zonation and Resultant Phenotypic Diversification Are Dictated by Blood Vessel Proximity. Cell Metab. 2019, 30, 201–211.e206. [Google Scholar] [CrossRef]
- Sun, Z.; Li, D.; Wu, H.; Hou, B. Tumour stem cell markers CD133 and CD44 are useful prognostic factors after surgical resection of pancreatic neuroendocrine tumours. Oncol. Lett. 2020, 20, 341. [Google Scholar] [CrossRef]
- Sun, Q.; Lesperance, J.; Wettersten, H.; Luterstein, E.; DeRose, Y.S.; Welm, A.; Cheresh, D.A.; Desgrosellier, J.S. Proapoptotic PUMA targets stem-like breast cancer cells to suppress metastasis. J. Clin. Investig. 2018, 128, 531–544. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Lei, R.; Zhuang, X.; Li, X.; Li, G.; Lev, S.; Segura, M.F.; Zhang, X.; Hu, G. MicroRNA-182 targets SMAD7 to potentiate TGFbeta-induced epithelial-mesenchymal transition and metastasis of cancer cells. Nat. Commun. 2016, 7, 13884. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Wells, A.; Yates, C.; Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Burkhalter, R.J.; Westfall, S.D.; Liu, Y.; Stack, M.S. Lysophosphatidic Acid Initiates Epithelial to Mesenchymal Transition and Induces beta-Catenin-mediated Transcription in Epithelial Ovarian Carcinoma. J. Biol. Chem. 2015, 290, 22143–22154. [Google Scholar] [CrossRef] [Green Version]
- Celia-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem. Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal 2009, 2, re3. [Google Scholar] [CrossRef] [Green Version]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem. Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [Green Version]
- Clezardin, P.; Frappart, L.; Clerget, M.; Pechoux, C.; Delmas, P.D. Expression of thrombospondin (TSP1) and its receptors (CD36 and CD51) in normal, hyperplastic, and neoplastic human breast. Cancer Res. 1993, 53, 1421–1430. [Google Scholar]
- Uray, I.P.; Liang, Y.; Hyder, S.M. Estradiol down-regulates CD36 expression in human breast cancer cells. Cancer Lett. 2004, 207, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martin, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Zhou, L.; Shen, T.; Zhou, S.; Ding, G.; Cao, L. Down-expression of CD36 in pancreatic adenocarcinoma and its correlation with clinicopathological features and prognosis. J. Cancer 2018, 9, 578–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem. Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buishand, F.O.; Arkesteijn, G.J.; Feenstra, L.R.; Oorsprong, C.W.; Mestemaker, M.; Starke, A.; Speel, E.J.; Kirpensteijn, J.; Mol, J.A. Identification of CD90 as Putative Cancer Stem Cell Marker and Therapeutic Target in Insulinomas. Stem. Cells Dev. 2016, 25, 826–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Su, Y.; Mei, Y.; Leng, Q.; Leng, B.; Liu, Z.; Stass, S.A.; Jiang, F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Investig. 2010, 90, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Marhaba, R.; Klingbeil, P.; Nuebel, T.; Nazarenko, I.; Buechler, M.W.; Zoeller, M. CD44 and EpCAM: Cancer-initiating cell markers. Curr. Mol. Med. 2008, 8, 784–804. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Herring, B.; Jang, S.; Whitt, J.; Goliwas, K.; Aburjania, Z.; Dudeja, V.; Ren, B.; Berry, J.; Bibb, J.; Frost, A.; et al. Ex Vivo Modeling of Human Neuroendocrine Tumors in Tissue Surrogates. Front. Endocrinol. 2021, 12, 710009. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Meyer-Rochow, G.Y.; Jackson, N.E.; Conaglen, J.V.; Whittle, D.E.; Kunnimalaiyaan, M.; Chen, H.; Westin, G.; Sandgren, J.; Stalberg, P.; Khanafshar, E.; et al. MicroRNA profiling of benign and malignant pheochromocytomas identifies novel diagnostic and therapeutic targets. Endocr. Relat. Cancer 2010, 17, 835–846. [Google Scholar] [CrossRef]
- Fendrich, V.; Maschuw, K.; Waldmann, J.; Buchholz, M.; Rehm, J.; Gress, T.M.; Bartsch, D.K.; Konig, A. Epithelial-mesenchymal transition is a critical step in tumorgenesis of pancreatic neuroendocrine tumors. Cancers 2012, 4, 281–294. [Google Scholar] [CrossRef] [Green Version]
- Tse, J.C.; Kalluri, R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J. Cell Biochem. 2007, 101, 816–829. [Google Scholar] [CrossRef]
- Vleminckx, K.; Vakaet, L., Jr.; Mareel, M.; Fiers, W.; van Roy, F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991, 66, 107–119. [Google Scholar] [CrossRef]
- Bracken, C.P.; Goodall, G.J. The many regulators of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2021, 23, 89–90. [Google Scholar] [CrossRef]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem. Cell 2015, 17, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Storz, P. Protein kinase D1: Gatekeeper of the epithelial phenotype and key regulator of cancer metastasis? Br. J. Cancer 2018, 118, 459–461. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Targeting Lysophosphatidic Acid in Cancer: The Issues in Moving from Bench to Bedside. Cancers 2019, 11, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Ramchandran, R.; Yang, X. Editorial: Molecular Mechanisms and Signaling in Endothelial Cell Biology and Vascular Heterogeneity. Front. Cell Dev. Biol. 2021, 9, 821100. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, B.; Deng, Y.; Mukhopadhyay, A.; Lanahan, A.A.; Zhuang, Z.W.; Moodie, K.L.; Mulligan-Kehoe, M.J.; Byzova, T.V.; Peterson, R.T.; Simons, M. ERK1/2-Akt1 crosstalk regulates arteriogenesis in mice and zebrafish. J. Clin. Investig. 2010, 120, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell Biol. 2005, 25, 8520–8530. [Google Scholar] [CrossRef] [Green Version]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis—Correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef]
- Zhou, B.; Xiang, J.; Jin, M.; Zheng, X.; Li, G.; Yan, S. High vimentin expression with E-cadherin expression loss predicts a poor prognosis after resection of grade 1 and 2 pancreatic neuroendocrine tumors. BMC Cancer 2021, 21, 334. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Jiang, Y.; Rose, J.B.; Nagaraju, G.P.; Jaskula-Sztul, R.; Hjelmeland, A.B.; Beck, A.W.; Chen, H.; Ren, B. Protein Kinase D1 Signaling in Cancer Stem Cells with Epithelial-Mesenchymal Plasticity. Cells 2022, 11, 3885. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233885
Guo Y, Jiang Y, Rose JB, Nagaraju GP, Jaskula-Sztul R, Hjelmeland AB, Beck AW, Chen H, Ren B. Protein Kinase D1 Signaling in Cancer Stem Cells with Epithelial-Mesenchymal Plasticity. Cells. 2022; 11(23):3885. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233885
Chicago/Turabian StyleGuo, Yichen, Yinan Jiang, J. Bart Rose, Ganji Purnachandra Nagaraju, Renata Jaskula-Sztul, Anita B. Hjelmeland, Adam W. Beck, Herbert Chen, and Bin Ren. 2022. "Protein Kinase D1 Signaling in Cancer Stem Cells with Epithelial-Mesenchymal Plasticity" Cells 11, no. 23: 3885. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11233885