
Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Metabolic Alterations in Cancer
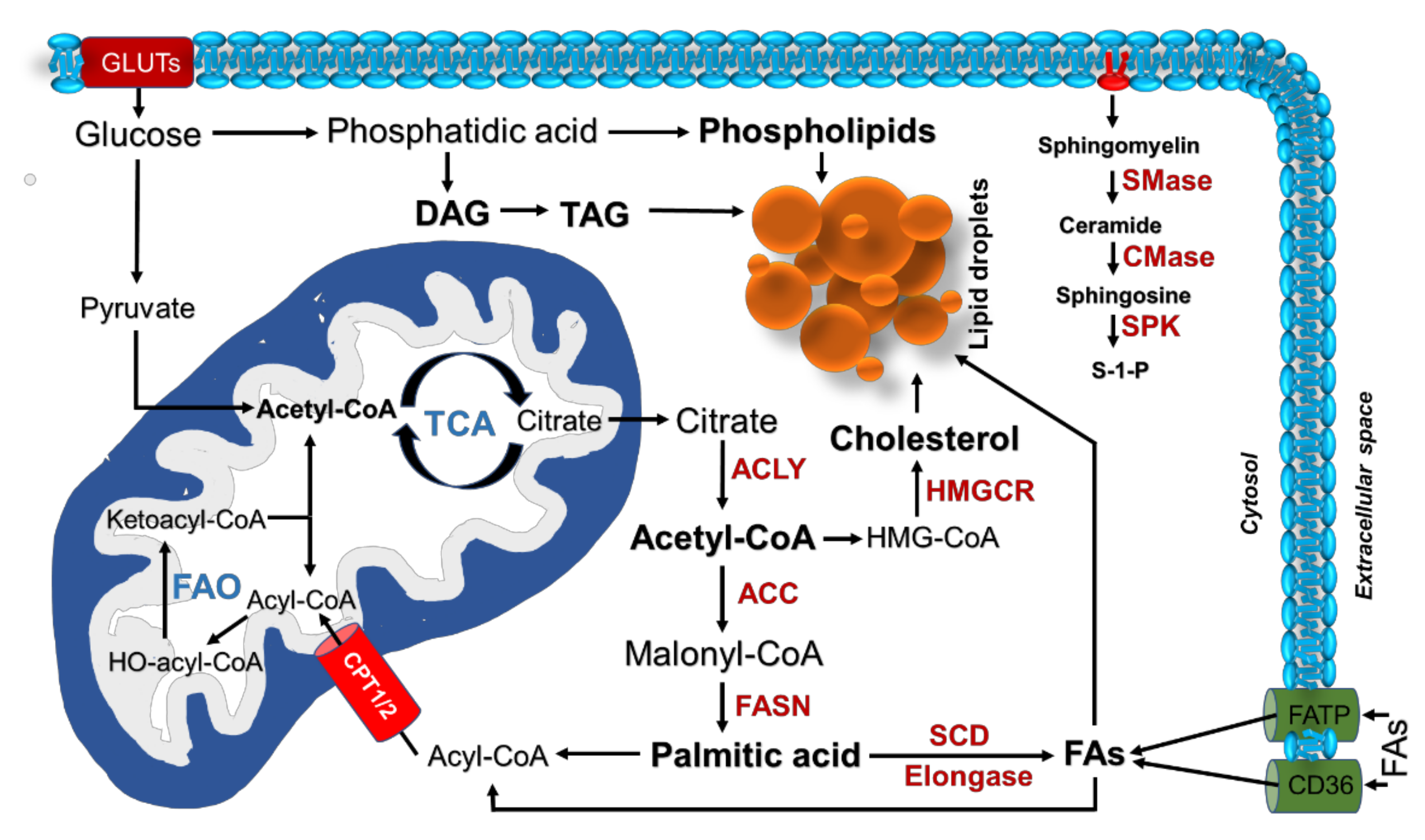
3. Alterations of Lipid Metabolism in Lung Cancer
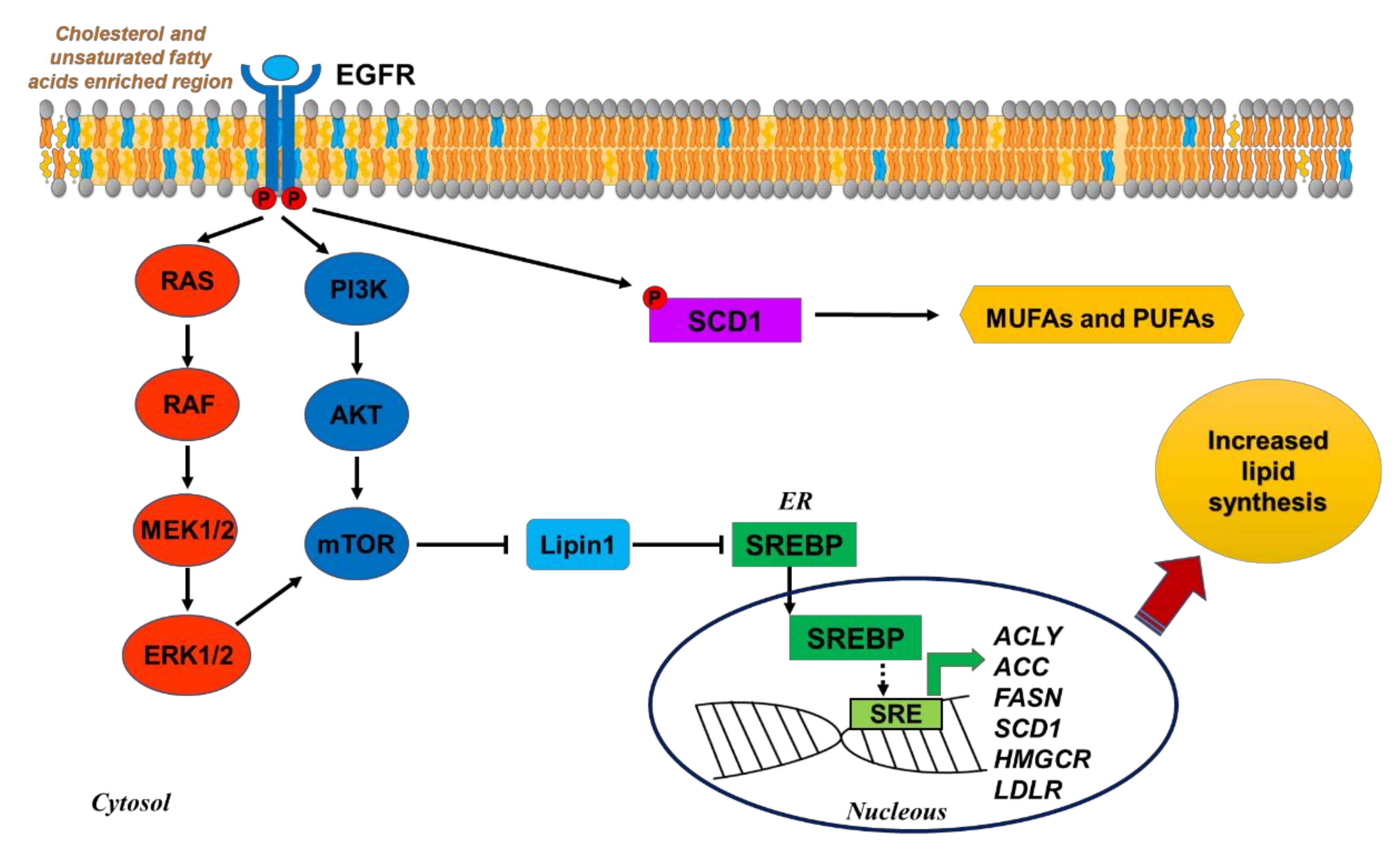
3.1. Fatty Acid Biosynthesis
3.2. Transport and Storage of Lipids
3.3. Fatty Acid Oxidation
3.4. Sphingolipid Metabolism
3.5. Cholesterol Metabolism
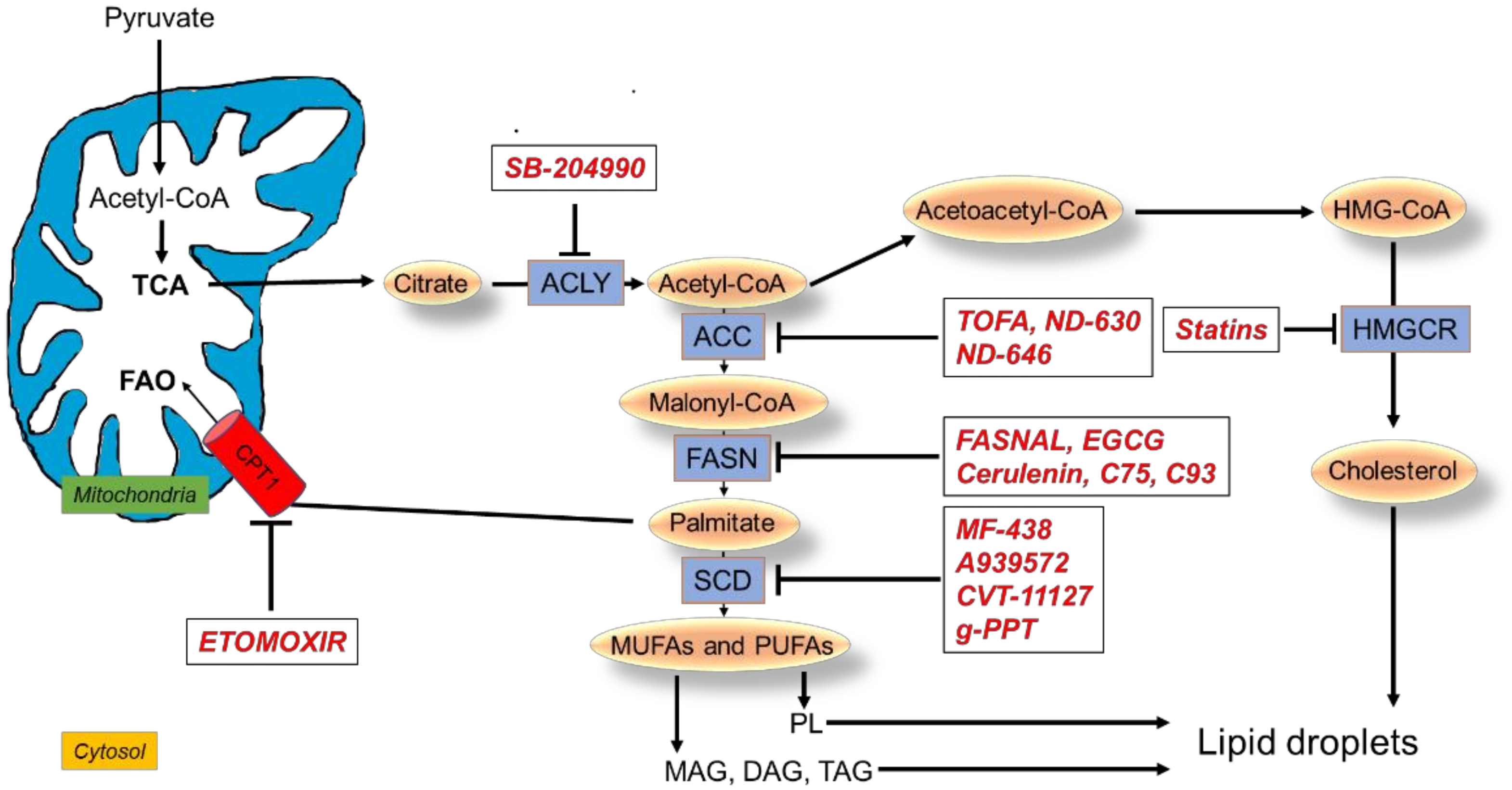
4. Targeting Lipid Metabolic Reprogramming in Lung Cancer
4.1. Targeting FA Synthesis
4.1.1. Inhibitors of ACLY
4.1.2. Inhibitors of ACC
4.1.3. Inhibitors of FASN
4.1.4. Inhibitors of SCD1
4.2. Targeting FAO
4.3. Targeting Cholesterol Metabolism
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- De Sousa, V.M.L.; Carvalho, L. Heterogeneity in Lung Cancer. Pathobiology 2018, 85, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Marrugal, Á.; Ojeda, L.; Paz-Ares, L.; Molina-Pinelo, S.; Ferrer, I. Proteomic-Based Approaches for the Study of Cytokines in Lung Cancer. Dis. Markers 2016, 2016, 2138627. [Google Scholar] [CrossRef] [Green Version]
- Chaitanya Thandra, K.; Barsouk, A.; Saginala, K.; Sukumar Aluru, J.; Barsouk, A. Epidemiology of Lung Cancer. Contemp. Oncol. 2021, 25, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Fang, S.; Gu, W. The Molecular Mechanism of Metabolic Remodeling in Lung Cancer. J. Cancer 2020, 11, 1403–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romaszko, A.; Doboszyńska, A. Multiple Primary Lung Cancer: A Literature Review. Adv. Clin. Exp. Med. 2018, 27, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Marien, E.; Meister, M.; Muley, T.; Fieuws, S.; Bordel, S.; Derua, R.; Spraggins, J.; van de Plas, R.; Dehairs, J.; Wouters, J.; et al. Non-small Cell Lung Cancer Is Characterized by Dramatic Changes in Phospholipid Profiles. Int. J. Cancer 2015, 137, 1539–1548. [Google Scholar] [CrossRef]
- Merino Salvador, M.; Gómez de Cedrón, M.; Moreno Rubio, J.; Falagán Martínez, S.; Sánchez Martínez, R.; Casado, E.; de Molina, A.R.; Sereno, M. Lipid Metabolism and Lung Cancer. Crit. Rev. Oncol. Hemat. 2017, 112, 31–40. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-Ras Mutation Subtypes in NSCLC and Associated Co-Occuring Mutations in Other Oncogenic Pathways. J. Thorac. Oncol. 2019, 14, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, 6487. [Google Scholar] [CrossRef]
- Torresano, L.; Nuevo-Tapioles, C.; Santacatterina, F.; Cuezva, J.M. Metabolic Reprogramming and Disease Progression in Cancer Patients. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165721. [Google Scholar] [CrossRef]
- Mendes, C.; Serpa, J. Metabolic Remodelling: An Accomplice for New Therapeutic Strategies to Fight Lung Cancer. Antioxidants 2019, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osugi, J.; Yamaura, T.; Muto, S.; Okabe, N.; Matsumura, Y.; Hoshino, M.; Higuchi, M.; Suzuki, H.; Gotoh, M. Prognostic Impact of the Combination of Glucose Transporter 1 and ATP Citrate Lyase in Node-Negative Patients with Non-Small Lung Cancer. Lung Cancer 2015, 88, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Freter, C. Lipid Metabolism, Apoptosis and Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 924–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, M.; Chen, S.; Lao, T.; Liang, D.; Sang, N. Nitrogen Anabolism Underlies the Importance of Glutaminolysis in Proliferating Cells. Cell Cycle 2010, 9, 3921–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, M.; Nakayama, K.I. A Second Warburg-like Effect in Cancer Metabolism: The Metabolic Shift of Glutamine-derived Nitrogen. BioEssays 2020, 42, e2000169. [Google Scholar] [CrossRef]
- Chen, W.-C.; Wang, C.-Y.; Hung, Y.-H.; Weng, T.-Y.; Yen, M.-C.; Lai, M.-D. Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar]
- Wang, J.; Li, Y. CD36 Tango in Cancer: Signaling Pathways and Functions. Theranostics 2019, 9, 4893–4908. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Menendez, J. Pharmacological Inhibitors of Fatty Acid Synthase (FASN)-Catalyzed Endogenous Fatty Acid Biogenesis: A New Family of Anti-Cancer Agents? Curr. Pharm. Biotechnol. 2006, 7, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, F.; Zhao, X.; Jiang, H.; Wu, X.; Wang, B.; Zhou, M.; Tian, M.; Shi, B.; Wang, H.; et al. EGFR Modulates Monounsaturated Fatty Acid Synthesis through Phosphorylation of SCD1 in Lung Cancer. Mol. Cancer 2017, 16, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid Metabolic Reprogramming in Cancer Cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Jin, C.; Yuan, P. Implications of Lipid Droplets in Lung Cancer: Associations with Drug Resistance. Oncol. Lett. 2020, 20, 2091–2104. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhou, J.; Hooi, S.; Jiang, Y.; Lu, G. Fatty Acid Activation in Carcinogenesis and Cancer Development: Essential Roles of Long chain Acyl CoA Synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.-Y.; Fang, X. Fatty Acid Oxidation: An Emerging Facet of Metabolic Transformation in Cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine Palmitoyltransferase 1C Promotes Cell Survival and Tumor Growth under Conditions of Metabolic Stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Vanhove, K.; Derveaux, E.; Graulus, G.-J.; Mesotten, L.; Thomeer, M.; Noben, J.-P.; Guedens, W.; Adriaensens, P. Glutamine Addiction and Therapeutic Strategies in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 252. [Google Scholar] [CrossRef] [Green Version]
- Fumarola, C.; Petronini, P.G.; Alfieri, R. Impairing energy metabolism in solid tumors through agents targeting oncogenic signaling pathways. Biochem Pharmacol. 2018, 151, 114–125. [Google Scholar] [CrossRef]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavareá, J.M. The Identification of ATP-Citrate Lyase as a Protein Kinase B (Akt) Substrate in Primary Adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef] [Green Version]
- Ricoult, S.J.H.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras Stimulate de Novo Lipid Synthesis through MTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Griffiths, B.; Chung, Y.-L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt Induces Transcription of Enzymes Involved in Cholesterol and Fatty Acid Biosynthesis via Activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid Metabolism Reprogramming and Its Potential Targets in Cancer. Cancer Commun. 2018, 38, 27. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The Interplay between Cell Signalling and the Mevalonate Pathway in Cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Gouw, A.M.; Margulis, K.; Liu, N.S.; Raman, S.J.; Mancuso, A.; Toal, G.G.; Tong, L.; Mosley, A.; Hsieh, A.L.; Sullivan, D.K.; et al. The MYC Oncogene Cooperates with Sterol-Regulated Element-Binding Protein to Regulate Lipogenesis Essential for Neoplastic Growth. Cell Metab. 2019, 30, 556–572. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid Metabolism in Cancer Progression and Therapeutic Strategies. MedComm 2021, 2, 27–59. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, X.; Song, G. The Hippo Pathway: A Master Regulatory Network Important in Cancer. Cells 2021, 10, 1416. [Google Scholar] [CrossRef]
- El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Mi, S.; Ye, J.; Lou, G. Aberrant Lipid Metabolism in Cancer Cells and Tumor Microenvironment: The Player Rather than Bystander in Cancer Progression and Metastasis. J. Cancer 2021, 12, 7498–7506. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, Y.; Mao, Q.; Fan, W.; Xu, L.; Chen, Y.; Xu, L.; Wang, J. FLOT1 Promotes Tumor Development, Induces Epithelial–Mesenchymal Transition, and Modulates the Cell Cycle by Regulating the Erk/Akt Signaling Pathway in Lung Adenocarcinoma. Thorac. Cancer 2019, 10, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP Citrate Lyase: Activation and Therapeutic Implications in Non–Small Cell Lung Cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP Citrate Lyase Inhibition Can Suppress Tumor Cell Growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Xin, M.; Qiao, Z.; Li, J.; Liu, J.; Song, S.; Zhao, X.; Miao, P.; Tang, T.; Wang, L.; Liu, W.; et al. MiR-22 Inhibits Tumor Growth and Metastasis by Targeting ATP Citrate Lyase: Evidence in Osteosarcoma, Prostate Cancer, Cervical Cancer and Lung Cancer. Oncotarget 2016, 7, 44252–44265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, E.-Q.; Zhao, W.; Zhang, C.; Qin, L.-Z.; Liu, S.-J.; Feng, Z.-Q.; Wen, X.; Chen, C.-P. Synthesis and Anti-Cancer Activity of ND-646 and Its Derivatives as Acetyl-CoA Carboxylase 1 Inhibitors. Eur. J. Pharm. Sci. 2019, 137, 105010. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The Acetyl-CoA Carboxylase Enzyme: A Target for Cancer Therapy? Expert Rev. Anticancer Ther. 2015, 15, 667–676. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Frost, A.R.; Manne, U.; Bell, W.C.; Weiss, H.; Heimburger, D.C.; Grizzle, W.E. The Expression of Fatty Acid Synthase (FASE) Is an Early Event in the Development and Progression of Squamous Cell Carcinoma of the Lung. Hum. Pathol. 2000, 31, 1068–1073. [Google Scholar] [CrossRef]
- Orita, H.; Coulter, J.; Tully, E.; Kuhajda, F.P.; Gabrielson, E. Inhibiting Fatty Acid Synthase for Chemoprevention of Chemically Induced Lung Tumors. Clin. Cancer Res. 2008, 14, 2458–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visca, P.; Sebastiani, V.; Botti, C.; Diodoro, M.G.; Lasagni, R.P.; Romagnoli, F.; Brenna, A.; de Joannon, B.C.; Donnorso, R.P.; Lombardi, G.; et al. Fatty Acid Synthase (FAS) Is a Marker of Increased Risk of Recurrence in Lung Carcinoma. Anticancer Res. 2004, 24, 4169–4173. [Google Scholar] [PubMed]
- Luo, D.; Xiao, H.; Dong, J.; Li, Y.; Feng, G.; Cui, M.; Fan, S. B7-H3 Regulates Lipid Metabolism of Lung Cancer through SREBP1-Mediated Expression of FASN. Biochem. Biophys. Res. Commun. 2017, 482, 1246–1251. [Google Scholar] [CrossRef]
- Ali, A.; Levantini, E.; Teo, J.T.; Goggi, J.; Clohessy, J.G.; Wu, C.S.; Chen, L.; Yang, H.; Krishnan, I.; Kocher, O.; et al. Fatty Acid Synthase Mediates EGFR Palmitoylation in EGFR Mutated Non-small Cell Lung Cancer. EMBO Mol. Med. 2018, 10, e8313. [Google Scholar] [CrossRef]
- Chen, Z.; Yu, D.; Owonikoko, T.K.; Ramalingam, S.S.; Sun, S.-Y. Induction of SREBP1 Degradation Coupled with Suppression of SREBP1-Mediated Lipogenesis Impacts the Response of EGFR Mutant NSCLC Cells to Osimertinib. Oncogene 2021, 40, 6653–6665. [Google Scholar] [CrossRef]
- Xu, C.; Zhang, L.; Wang, D.; Jiang, S.; Cao, D.; Zao, Z.; Huang, M.; Jin, J. Lipidomics reveals that sustained SREBP-1-dependent lipogenesis is a key mediator of gefitinib-acquired resistance in EGFR-mutant lung cancer. Cell Death Discov. 2021, 7, 353. [Google Scholar] [CrossRef]
- Chang, L.; Fang, S.; Chen, Y.; Yang, Z.; Yuan, Y.; Zhang, J.; Ye, L.; Gu, W. Inhibition of FASN Suppresses the Malignant Biological Behavior of Non-Small Cell Lung Cancer Cells via Deregulating Glucose Metabolism and AKT/ERK Pathway. Lipids Health Dis. 2019, 18, 118. [Google Scholar] [CrossRef] [Green Version]
- Zhan, N.; Li, B.; Xu, X.; Xu, J.; Hu, S. Inhibition of FASN Expression Enhances Radiosensitivity in Human Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 15, 4578–4584. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Fan, X.-X.; He, J.; Pan, H.; Li, R.-Z.; Huang, L.; Jiang, Z.; Yao, X.-J.; Liu, L.; Lai-Han Leung, E.; et al. SCD1 Is Associated with Tumor Promotion, Late Stage and Poor Survival in Lung Adenocarcinoma. Oncotarget 2016, 7, 39970–39979. [Google Scholar] [CrossRef] [Green Version]
- Noto, A.; Raffa, S.; de Vitis, C.; Roscilli, G.; Malpicci, D.; Coluccia, P.; di Napoli, A.; Ricci, A.; Giovagnoli, M.R.; Aurisicchio, L.; et al. Stearoyl-CoA Desaturase-1 Is a Key Factor for Lung Cancer-Initiating Cells. Cell Death Dis. 2013, 4, e947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noto, A.; de Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-Desaturase 1 Regulates Lung Cancer Stemness via Stabilization and Nuclear Localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Nashed, M.; Chisholm, J.W.; Igal, R.A. Stearoyl-CoA Desaturase Activity Modulates the Activation of Epidermal Growth Factor Receptor in Human Lung Cancer Cells. Exp. Biol. Med. 2012, 237, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.S.; Yip, L.Y.; Basri, N.; Chong, V.S.H.; Teo, C.C.; Tan, E.; Lim, K.L.; Tan, G.S.; Yang, X.; Yeo, S.Y.; et al. Lipidomic Profiling of Lung Pleural Effusion Identifies Unique Metabotype for EGFR Mutants in Non-Small Cell Lung Cancer. Sci. Rep. 2016, 6, 35110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglia, N.; Igal, R.A. Inhibition of Stearoyl-CoA Desaturase 1 Expression in Human Lung Adenocarcinoma Cells Impairs Tumorigenesis. Int. J. Oncol. 2008, 33, 839–850. [Google Scholar]
- Ni, K.; Wang, D.; Xu, H.; Mei, F.; Wu, C.; Liu, Z.; Zhou, B. MiR-21 Promotes Non-Small Cell Lung Cancer Cells Growth by Regulating Fatty Acid Metabolism. Cancer Cell Int. 2019, 19, 219. [Google Scholar] [CrossRef] [Green Version]
- McKillop, I.H.; Girardi, C.A.; Thompson, K.J. Role of Fatty Acid Binding Proteins (FABPs) in Cancer Development and Progression. Cell Signal. 2019, 62. [Google Scholar] [CrossRef]
- Tang, Z.; Shen, Q.; Xie, H.; Zhou, X.; Li, J.; Feng, J.; Liu, H.; Wang, W.; Zhang, S.; Ni, S. Elevated Expression of FABP3 and FABP4 Cooperatively Correlates with Poor Prognosis in Non-Small Cell Lung Cancer (NSCLC). Oncotarget 2016, 7, 46253–46262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Wang, Q.; Li, D.; Wei, X.; Jia, Y.; Zhang, Z.; Ai, B.; Cao, X.; Guo, T.; Liao, Y. Co-Administration of 20(S)-Protopanaxatriol (g-PPT) and EGFR-TKI Overcomes EGFR-TKI Resistance by Decreasing SCD1 Induced Lipid Accumulation in Non-Small Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 129. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, K.; Xu, Z.; Cui, G.; Zhang, X. Integrated Omics and Gene Expression Analysis Identifies the Loss of Metabolite-Metabolite Correlations in Small Cell Lung Cancer. Onco. Targets Ther. 2018, 11, 3919–3929. [Google Scholar] [CrossRef] [Green Version]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, S.M.; Chandel, N.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar]
- Li, F.; Han, X.; Li, F.; Wang, R.; Wang, H.; Gao, Y.; Wang, X.; Fang, Z.; Zhang, W.; Yao, S.; et al. LKB1 Inactivation Elicits a Redox Imbalance to Modulate Non-small Cell Lung Cancer Plasticity and Therapeutic Response. Cancer Cell. 2015, 27, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef]
- Li, S.; Zhu, X.; Wang, L.; Zheng, Z. The Role of Radiation Dose-Dependent Enhancement of Fatty Acid Oxidation in Radiation Surviving/Resistant Lung Cancer Cells. Journal of Clinical Oncology 2020, 38, e21724. [Google Scholar] [CrossRef]
- Li, J.; Zhao, S.; Zhou, X.; Zhang, T.; Zhao, L.; Miao, P.; Song, S.; Sun, X.; Liu, J.; Zhao, X.; et al. Inhibition of Lipolysis by Mercaptoacetate and Etomoxir Specifically Sensitize Drug-Resistant Lung Adenocarcinoma Cell to Paclitaxel. PLoS ONE 2013, 8, e74623. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and Cancer: Ceramide and Sphingosine-1-Phosphate in the Regulation of Cell Death and Drug Resistance. Future Oncol. 2010, 6, 1603–1624. [Google Scholar] [CrossRef] [Green Version]
- Goldkorn, T.; Chung, S.; Filosto, S. Lung Cancer and Lung Injury: The Dual Role of Ceramide. Handb Exp. Pharmacol. 2013, 216, 93–113. [Google Scholar]
- Suzuki, M.; Cao, K.; Kato, S.; Mizutani, N.; Tanaka, K.; Arima, C.; Tai, M.C.; Nakatani, N.; Yanagisawa, K.; Takeuchi, T.; et al. CERS6 Required for Cell Migration and Metastasis in Lung Cancer. J. Cell. Mol. Med. 2020, 24, 11949–11959. [Google Scholar] [CrossRef]
- Dai, L.; Smith, C.D.; Foroozesh, M.; Miele, L.; Qin, Z. The Sphingosine Kinase 2 Inhibitor ABC294640 Displays Anti-non-small Cell Lung Cancer Activities in Vitro and in Vivo. Int. J. Cancer 2018, 142, 2153–2162. [Google Scholar] [CrossRef] [Green Version]
- Alberg, A.J.; Armeson, K.; Pierce, J.S.; Bielawski, J.; Bielawska, A.; Visvanathan, K.; Hill, E.G.; Ogretmen, B. Plasma Sphingolipids and Lung Cancer: A Population-Based, Nested Case–Control Study. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 1374–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, S.; Aoki, K.; Mizutani, T.; Amano, M.; Nishimura, S.-I.; Haga, H. Glycosphingolipid GM2 Induces Invasiveness in Irradiation-Tolerant Lung Cancer Cells. Cell Struct. Funct. 2018, 43, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyler, A.; Johansson, A.; Karlsson, T.; Gudey, S.K.; Brännström, T.; Grankvist, K.; Behnam-Motlagh, P. Targeting Glucosylceramide Synthase Induction of Cell Surface Globotriaosylceramide (Gb3) in Acquired Cisplatin-Resistance of Lung Cancer and Malignant Pleural Mesothelioma Cells. Exp. Cell Res. 2015, 336, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravid, T.; Tsaba, A.; Gee, P.; Rasooly, R.; Medina, E.A.; Goldkorn, T. Ceramide Accumulation Precedes Caspase-3 Activation during Apoptosis of A549 Human Lung Adenocarcinoma Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L1082–L1092. [Google Scholar] [CrossRef] [Green Version]
- Kurinna, S.M.; Tsao, C.C.; Nica, A.F.; Jiffar, T.; Ruvolo, P.P. Ceramide Promotes Apoptosis in Lung Cancer-Derived A549 Cells by a Mechanism Involving c-Jun NH 2 -Terminal Kinase. Cancer Res. 2004, 64, 7852–7856. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Fong, Y.; Tsai, E.-M.; Chang, Y.-G.; Chou, H.; Wu, C.-Y.; Teng, Y.-N.; Liu, T.-C.; Yuan, S.-S.; Chiu, C.-C. Exogenous C8-Ceramide Induces Apoptosis by Overproduction of ROS and the Switch of Superoxide Dismutases SOD1 to SOD2 in Human Lung Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3010. [Google Scholar] [CrossRef] [Green Version]
- Tamanoi, F.; Azizian, M.; Ashrafi, M.; Bathaie, S. Mevalonate Pathway and Human Cancers. Curr. Mol. Pharmacol. 2017, 10, 77–85. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Jan, Y.-H.; Liu, Y.-P.; Yang, C.-J.; Su, C.-Y.; Chang, Y.-C.; Lai, T.-C.; Chiou, J.; Tsai, H.-Y.; Lu, J.; et al. Squalene Synthase Induces Tumor Necrosis Factor Receptor 1 Enrichment in Lipid Rafts to Promote Lung Cancer Metastasis. Am. J. Respir. Crit. Care Med. 2014, 190, 675–687. [Google Scholar] [CrossRef]
- Li, J.; Yan, H.; Zhao, L.; Jia, W.; Yang, H.; Liu, L.; Zhou, X.; Miao, P.; Sun, X.; Song, S.; et al. Inhibition of SREBP Increases Gefitinib Sensitivity in Non-Small Cell Lung Cancer Cells. Oncotarget 2016, 7, 52392–52403. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Yang, Y.; Peng, P.; Zhan, J.; Wang, Z.; Zhu, Z.; Zhang, Z.; Liu, L.; Fang, W.; Zhang, L. Cholesterol Synthesis Disruption Combined with a Molecule-Targeted Drug Is a Promising Metabolic Therapy for EGFR Mutant Non-Small Cell Lung Cancer. Transl. Lung Cancer Res. 2021, 10, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Hall, Z.; Wilson, C.H.; Burkhart, D.L.; Ashmore, T.; Evan, G.I.; Griffin, J.L. Myc Linked to Dysregulation of Cholesterol Transport and Storage in Nonsmall Cell Lung Cancer. J. Lipid Res. 2020, 61, 1390–1399. [Google Scholar] [CrossRef] [PubMed]
- Shares, B.H.; Busch, M.; White, N.; Shum, L.; Eliseev, R.A. Active Mitochondria Support Osteogenic Differentiation by Stimulating β-Catenin Acetylation. J. Biol. Chem. 2018, 293, 16019–16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, J.; Huang, G.; Zhao, Y.; Yue, X.; Wu, H.; Li, J.; Zhu, J.; Shen, Z.; Haffty, B.G.; et al. Cullin3–KLHL25 Ubiquitin Ligase Targets ACLY for Degradation to Inhibit Lipid Synthesis and Tumor Progression. Genes Dev. 2016, 30, 1956–1970. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xu, C.; Sun, M.; Luo, D.; Liao, D.; Cao, D. Acetyl-CoA Carboxylase-α Inhibitor TOFA Induces Human Cancer Cell Apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Qiu, L.; Wu, B.; Shen, H.; Zhu, J.; Zhou, L.; Gu, L.; Di, W. TOFA Suppresses Ovarian Cancer Cell Growth in Vitro and in Vivo. Mol. Med. Rep. 2013, 8, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Guseva, N.V.; Rokhlin, O.W.; Glover, R.A.; Cohen, M.B. TOFA (5-Tetradecyl-Oxy-2-Furoic Acid) Reduces Fatty Acid Synthesis, Inhibits Expression of AR, Neuropilin-1 and Mcl-1 and Kills Prostate Cancer Cells Independent of P53 Status. Cancer Bio. Ther. 2011, 12, 80–85. [Google Scholar] [CrossRef] [Green Version]
- Harriman, G.; Greenwood, J.; Bhat, S.; Huang, X.; Wang, R.; Paul, D.; Tong, L.; Saha, A.K.; Westlin, W.F.; Kapeller, R.; et al. Acetyl-CoA Carboxylase Inhibition by ND-630 Reduces Hepatic Steatosis, Improves Insulin Sensitivity, and Modulates Dyslipidemia in Rats. Proc. Natl Acad. Sci. 2016, 113, E1796–E1805. [Google Scholar] [CrossRef] [Green Version]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of Acetyl-CoA Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of Non-Small-Cell Lung Cancer in Preclinical Models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, V.; Khayati, K.; Hu, Z.S.; Lee, A.; Kamran, W.; Su, X.; Guo, J.Y. Autophagy Modulates Lipid Metabolism to Maintain Metabolic Flexibility for Lkb1 -Deficient Kras -Driven Lung Tumorigenesis. Genes Dev. 2019, 33, 150–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relat, J.; Blancafort, A.; Oliveras, G.; Cufí, S.; Haro, D.; Marrero, P.F.; Puig, T. Different Fatty Acid Metabolism Effects of (−)-Epigallocatechin-3-Gallate and C75 in Adenocarcinoma Lung Cancer. BMC Cancer 2012, 12, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polonio-Alcalá, E.; Palomeras, S.; Torres-Oteros, D.; Relat, J.; Planas, M.; Feliu, L.; Ciurana, J.; Ruiz-Martínez, S.; Puig, T. Fatty Acid Synthase Inhibitor G28 Shows Anticancer Activity in EGFR Tyrosine Kinase Inhibitor Resistant Lung Adenocarcinoma Models. Cancers 2020, 12, 1283. [Google Scholar] [CrossRef]
- Tan, Y.-J.; Ali, A.; Tee, S.-Y.; Teo, J.-T.; Xi, Y.; Go, M.-L.; Lam, Y. Galloyl Esters of Trans-Stilbenes Are Inhibitors of FASN with Anticancer Activity on Non-Small Cell Lung Cancer Cells. Eur. J. Med. Chem. 2019, 182, 111597. [Google Scholar] [CrossRef]
- Bitencourt, T.A.; Komoto, T.T.; Massaroto, B.G.; Miranda, C.E.S.; Beleboni, R.O.; Marins, M.; Fachin, A.L. Trans-Chalcone and Quercetin down-Regulate Fatty Acid Synthase Gene Expression and Reduce Ergosterol Content in the Human Pathogenic Dermatophyte Trichophyton Rubrum. BMC Complement. Altern. Med. 2013, 13, 229. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; Vellon, L.; Cuyàs, E.; Buxó, M.; Martin-Castillo, B.; Serra, D.; García, J.; Lupu, R.; Menendez, J.A. Clinical and Therapeutic Relevance of the Metabolic Oncogene Fatty Acid Synthase in HER2+ Breast Cancer. Histol. Histopathol. 2017, 32, 687–698. [Google Scholar]
- Orita, H.; Coulter, J.; Lemmon, C.; Tully, E.; Vadlamudi, A.; Medghalchi, S.M.; Kuhajda, F.P.; Gabrielson, E. Selective Inhibition of Fatty Acid Synthase for Lung Cancer Treatment. Clin. Cancer Res. 2007, 13, 7139–7145. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, C.R.M.; Woo, J.-H.; Tully, E.; Wilsbach, K.; Gabrielson, E. Nuclear Factor-ΚB (NF-ΚB) Mediates a Protective Response in Cancer Cells Treated with Inhibitors of Fatty Acid Synthase. J. Biol. Chem. 2011, 286, 31457–31465. [Google Scholar] [CrossRef] [Green Version]
- Léger, S.; Black, W.C.; Deschenes, D.; Dolman, S.; Falgueyret, J.-P.; Gagnon, M.; Guiral, S.; Huang, Z.; Guay, J.; Leblanc, Y.; et al. Synthesis and Biological Activity of a Potent and Orally Bioavailable SCD Inhibitor (MF-438). Bioorganic Med. Chem. Lett. 2010, 20, 499–502. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Noto, A.; de Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-Desaturase 1 Activity Reverts Resistance to Cisplatin in Lung Cancer Stem Cells. Cancer Lett. 2017, 406, 93–104. [Google Scholar] [CrossRef]
- Xin, Z.; Zhao, H.; Serby, M.D.; Liu, B.; Liu, M.; Szczepankiewicz, B.G.; Nelson, L.T.J.; Smith, H.T.; Suhar, T.S.; Janis, R.S.; et al. Discovery of Piperidine-Aryl Urea-Based Stearoyl-CoA Desaturase 1 Inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4298–4302. [Google Scholar] [CrossRef] [PubMed]
- She, K.; Fang, S.; Du, W.; Fan, X.; He, J.; Pan, H.; Huang, L.; He, P.; Huang, J. SCD1 Is Required for EGFR-Targeting Cancer Therapy of Lung Cancer via Re-Activation of EGFR/PI3K/AKT Signals. Cancer Cell Int. 2019, 19, 103. [Google Scholar] [CrossRef]
- Hess, D.; Chisholm, J.W.; Igal, R.A. Inhibition of StearoylCoA Desaturase Activity Blocks Cell Cycle Progression and Induces Programmed Cell Death in Lung Cancer Cells. PLoS ONE 2010, 5, e11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglia, N.; Chisholm, J.W.; Igal, R.A. Inhibition of StearoylCoA Desaturase-1 Inactivates Acetyl-CoA Carboxylase and Impairs Proliferation in Cancer Cells: Role of AMPK. PLoS ONE 2009, 4, e6812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheeraj, A.; Agarwal, C.; Schlaepfer, I.R.; Raben, D.; Singh, R.; Agarwal, R.; Deep, G. A novel approach to target hypoxic cancer cells via combining β-oxidation inhibitor etomoxir with radiation. Hypoxia 2018, 6, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.-K.; Hu, Z.-G.; Tian, Y.-F.; Zeng, F.-J. Statin Use and Prognosis of Lung Cancer: A Systematic Review and Meta-Analysis of Observational Studies and Randomized Controlled Trials. Drug Des. Devel. Ther. 2019, 13, 405–422. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, X.; Zhang, R.; Xia, Y.; Shao, Z.; Mei, Z. Effects of Statin Exposure and Lung Cancer Survival: A Meta-Analysis of Observational Studies. Pharmacol. Res. 2019, 141, 357–365. [Google Scholar] [CrossRef]
- Fiala, O.; Pesek, M.; Finek, J.; Minarik, M.; Benesova, L.; Bortlicek, Z.; Topolcan, O. Statins Augment Efficacy of EGFR-TKIs in Patients with Advanced-Stage Non-Small Cell Lung Cancer Harbouring KRAS Mutation. Tumour Biol. 2015, 36, 5801–5805. [Google Scholar] [CrossRef]
- Hung, M.-S.; Chen, I.-C.; Lee, C.-P.; Huang, R.-J.; Chen, P.-C.; Tsai, Y.-H.; Yang, Y.-H. Statin Improves Survival in Patients with EGFR-TKI Lung Cancer: A Nationwide Population-Based Study. PLoS ONE 2017, 12, e0171137. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Lee, S.H.; Yoo, N.J.; Hyung, L.S.; Moon, Y.J.; Yun, T.; Kim, H.T.; Lee, J.S. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2011, 17, 1553–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, P.A.; Chang, C.C.; Galvin, C.J.; Wang, Y.C.; An, S.Y.; Huang, C.W.; Wang, Y.H.; Hsu, M.H.; Li, Y.J.; Yang, H.C. Statins use and its impact in EGFR-TKIs resistance to prolong the survival of lung cancer patients: A cancer registry cohort study in Taiwan. Cancer Sci. 2020, 111, 2965. [Google Scholar] [CrossRef] [PubMed]
- Youngjoo, L. Randomized Phase II Study of Afatinib Plus Simvastatin Versus Afatinib Alone in Previously Treated Patients with Advanced Nonadenocarcinomatous Non-small Cell Lung Cancer. Cancer Res Treat. 2017, 49, 1001–1011. [Google Scholar]
- Hanai, J.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of Lung Cancer Growth: ATP Citrate Lyase Knockdown and Statin Treatment Leads to Dual Blockade of Mitogen-Activated Protein Kinase (MAPK) and Phosphatidylinositol-3-Kinase (PI3K)/AKT Pathways. J. Cell. Physiol. 2012, 227, 1709–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitroulakos, J.; Lorimer, I.A.; Goss, G. Strategies to Enhance Epidermal Growth Factor Inhibition: Targeting the Mevalonate Pathway. Clin. Cancer Res. 2006, 12, 4426s–4431s. [Google Scholar] [CrossRef] [Green Version]
- Walther, U.; Emmrich, K.; Ramer, R.; Mittag, N.; Hinz, B. Lovastatin Lactone Elicits Human Lung Cancer Cell Apoptosis via a COX-2/PPARγ-Dependent Pathway. Oncotarget 2016, 7, 10345–10362. [Google Scholar] [CrossRef] [Green Version]
- Sanli, T.; Liu, C.; Rashid, A.; Hopmans, S.N.; Tsiani, E.; Schultz, C.; Farrell, T.; Singh, G.; Wright, J.; Tsakiridis, T. Lovastatin Sensitizes Lung Cancer Cells to Ionizing Radiation: Modulation of Molecular Pathways of Radioresistance and Tumor Suppression. J. Thorac. Oncol. 2011, 6, 439–450. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Bai, R.; Wang, Q.; Wang, K.; Li, X.; Liu, K.; Ryu, J.; Wang, T.; Chang, X.; Ma, W.; et al. Fluvastatin Inhibits HMG-CoA Reductase and Prevents Non–Small Cell Lung Carcinogenesis. Cancer Prev. Res. 2019, 12, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.Y.; Kim, I.K.; Lee, H.I.; Mo, J.Y.; Yeo, C.D.; Kang, H.H.; Moon, H.S.; Lee, S.H. The Apoptotic Effect of Simvastatin via the Upregulation of BIM in Nonsmall Cell Lung Cancer Cells. Exp. Lung Res. 2016, 42, 14–23. [Google Scholar] [CrossRef]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an Alternative Fatty Acid Desaturation Pathway Increasing Cancer Plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Cheng, T.; Zhang, J.; Liu, D.; Lai, G.; Wen, X. Prognosis of Non-Small-Cell Lung Cancer Patients with Lipid Metabolism Pathway Alternations to Immunotherapy. Front. Genet. 2021, 12, 646362. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Lipid Metabolism Alteration | EGFR-TKI | Overcoming Resistance | Refs |
|---|---|---|---|
| FASN-mediated palmitoylation of mutant EGFR | Gefitinib | Orlistat treatment | [48] |
| Elevated levels of SREBP1 and/or FASN | Osimertinib | Genetic knockdown of SREBP1 | [49] |
| FASN overexpression | Gefitinib/ Osimertinib | G28 treatment | [94] |
| Constitutive activation of SREBP1 | Gefitinib | Fatostatin treatment | [50] |
| High LD content and SCD1 expression | Gefitinib | Inhibition of LDs formation by g-PPT | [62] |
| FAO upregulation | Osimertinib | Etomoxir treatment | [67] |
| LDLR expression upregulation | Gefitinib | Atorvastatin treatment | [83] |
| Undefined | Gefitinib | Simvastatin treatment | [119] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eltayeb, K.; La Monica, S.; Tiseo, M.; Alfieri, R.; Fumarola, C. Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer. Cells 2022, 11, 413. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030413
Eltayeb K, La Monica S, Tiseo M, Alfieri R, Fumarola C. Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer. Cells. 2022; 11(3):413. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030413
Chicago/Turabian StyleEltayeb, Kamal, Silvia La Monica, Marcello Tiseo, Roberta Alfieri, and Claudia Fumarola. 2022. "Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer" Cells 11, no. 3: 413. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030413