
Mechanisms of Cisplatin Resistance in HPV Negative Head and Neck Squamous Cell Carcinomas

Abstract
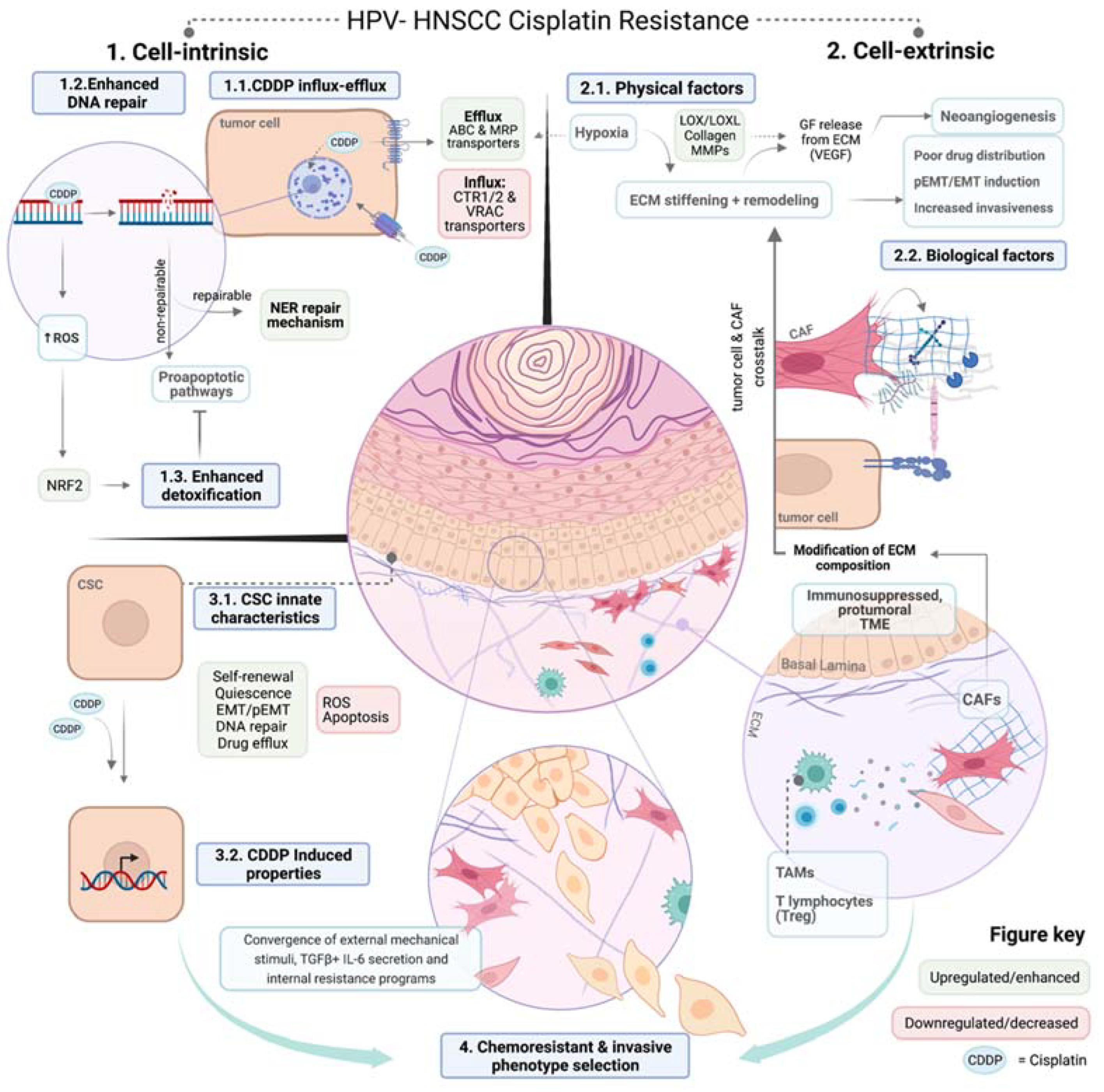
:1. Head and Neck Squamous Cell Carcinomas
1.1. Molecular Alterations of HVP Negative HNSCCs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affected Gene/Pathway | Type of Alteration | Frequency | Effect on Cell | Ref. |
|---|---|---|---|---|
| TP53 | Inactivation | 84% | Increased cell proliferation and apoptosis evasion | [9] |
| CDKN2A | Loss of function/ Deletion | 58% | [9] | |
| CASP8 | Inactivation | 11% | Apoptosis evasion | [9] |
| PIK3CA | Gain of function/Amplifications | 34% | Increased proliferation and pro-survival signals | [9] |
| MYC | 14% | [9] | ||
| CCND1 | 31% | [9] | ||
| Tyrosine kinase receptors | 60% | [9] | ||
| TP63 | Copy number gain | 19% | Pro-survival signals and cell differentiation inhibition | [9,11,12,13] |
| SOX2 | n.d. | [11,12,13] | ||
| NOTCH pathway | Inhibition | 26% | [15] | |
| FAT1 | Loss of function | 25–30% | Promotion of tumor progression and cancer stemness | [16] |
| NRF2/KEAP1 pathway | Activation | 20% | Increased celular detoxification | [17] |
1.2. Available Therapies for HNSCCs
1.3. Cisplatin Mechanisms of Action
2. Classical Mechanisms of Cisplatin Resistance
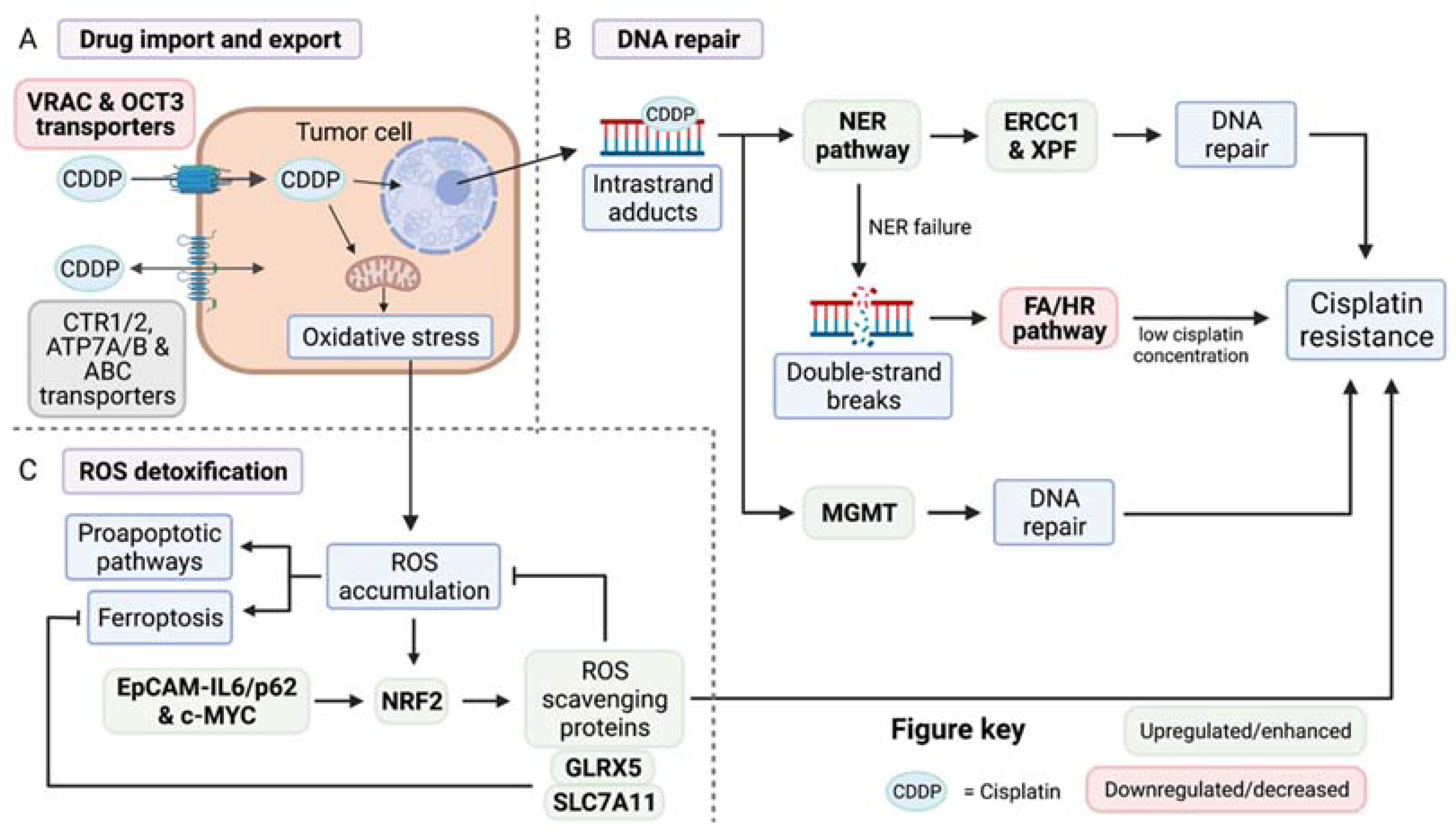
2.1. Control of Cisplatin Import and Export
2.2. Mechanisms of DNA Repair
2.3. Cellular Detoxification of Reactive Oxygen Species
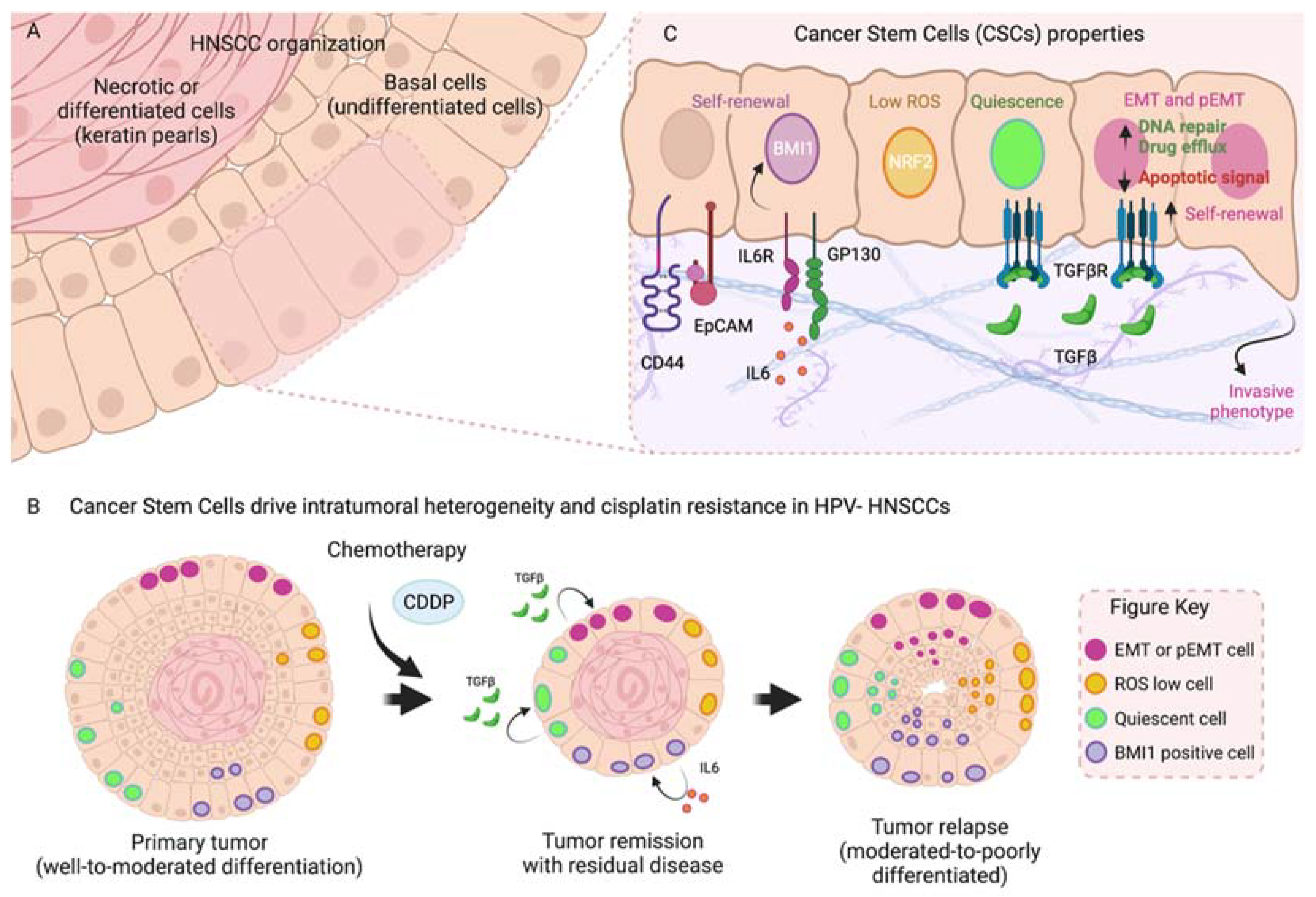
3. Tumor Heterogeneity: Cancer Stem Cells and Therapy Resistance
4. Epithelial to Mesenchymal Transition and Cell Plasticity
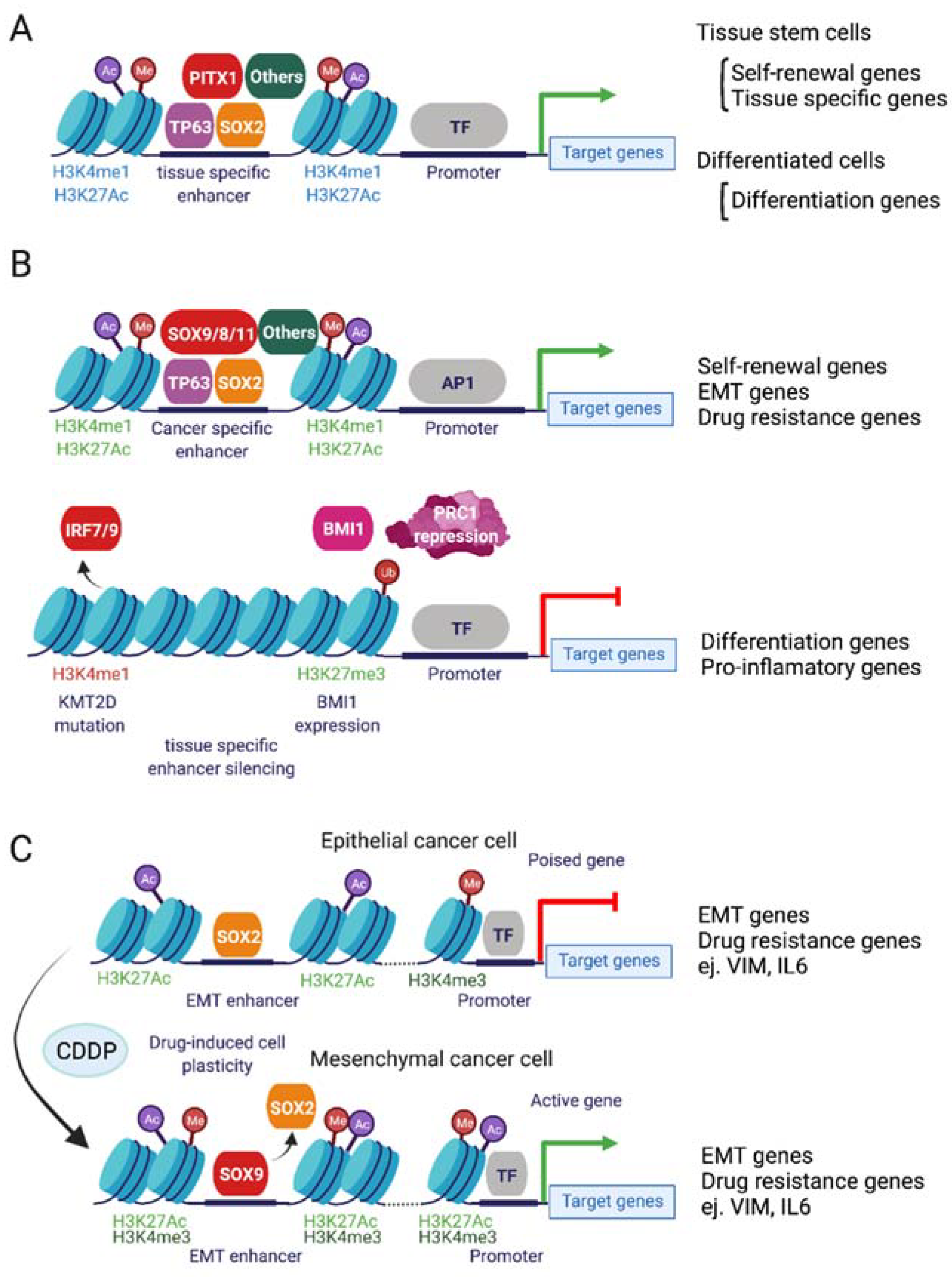
5. Epigenetic Mechanisms of Cisplatin Resistance
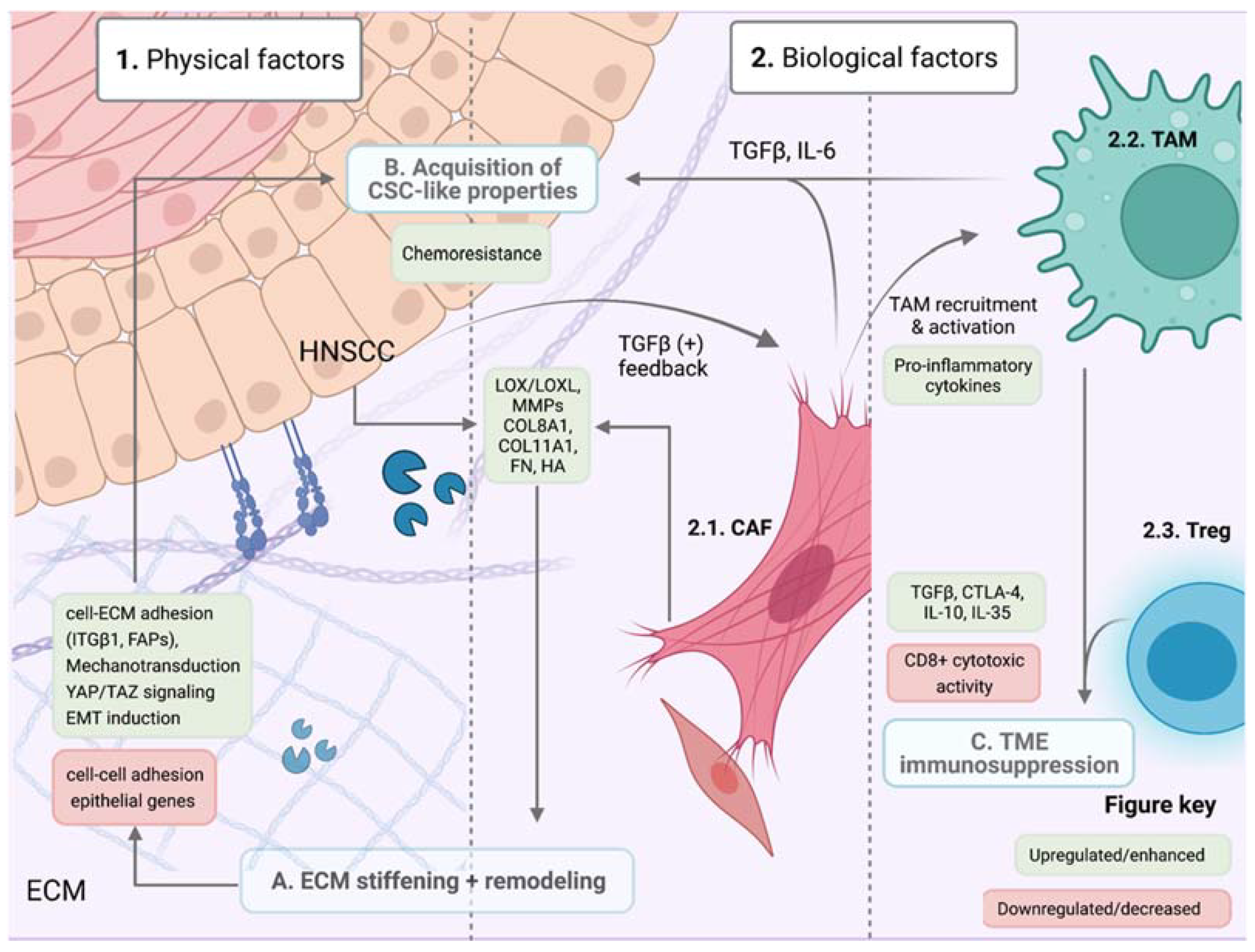
6. The Tumor Microenvironment and Therapy Resistance
6.1. Physical Factors: ECM Remodelling, Composition and Stiffness
6.2. Biological Factors
6.2.1. Cancer-Associated Fibroblasts
6.2.2. Tumor-Associated Macrophages and Lymphocytes
7. Other Pathways Involved in Cisplatin Resistance
7.1. YAP/TAZ Pathway
7.2. Survival Pathways: Regulation of STAT3 Pathway
7.3. Notch Pathway
7.4. Autophagy
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and Neck Squamous Cell Carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Michaud, D.S.; Langevin, S.M.; Eliot, M.; Nelson, H.H.; Pawlita, M.; McClean, M.D.; Kelsey, K.T. High-risk HPV Types and Head and Neck Cancer. Int. J. Cancer 2014, 135, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. List of Classifications by Cancer Sites with Sufficient or Limited Evidence in Humans, Volumes 1 to 127. IARC Monographs on the Identification of Carcinogenic Hazards to Humans. IARC. 2020. Available online: https://monographs.iarc.who.int/agents-classified-by-the-iarc/ (accessed on 1 February 2022).
- Isayeva, T.; Li, Y.; Maswahu, D.; Brandwein-Gensler, M. Human Papillomavirus in Non-Oropharyngeal Head and Neck Cancers: A Systematic Literature Review. Head Neck Pathol. 2012, 6, 104–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, A.P.; Saha, S.; Kraninger, J.L.; Swick, A.D.; Yu, M.; Lambert, P.F.; Kimple, R.J. Prevalence of Human Papillomavirus in Oropharyngeal Cancer: A Systematic Review. Cancer J. Sudbury Mass 2015, 21, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whang, S.N.; Filippova, M.; Duerksen-Hughes, P. Recent Progress in Therapeutic Treatments and Screening Strategies for the Prevention and Treatment of HPV-Associated Head and Neck Cancer. Viruses 2015, 7, 5040–5065. [Google Scholar] [CrossRef] [Green Version]
- Syrjänen, S.; Rautava, J. HPV Infection in Head and Neck Cancer. Recent Results Cancer 2016, 257–267. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive Genomic Characterization of Head and Neck Squamous Cell Carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Pendleton, K.P.; Grandis, J.R. Cisplatin-Based Chemotherapy Options for Recurrent and/or Metastatic Squamous Cell Cancer of the Head and Neck. Clin. Med. Insights Ther. 2013, 5, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, M.R.; Wilson, C.; Ory, B.; Rothenberg, S.M.; Faquin, W.; Mills, A.A.; Ellisen, L.W. FGFR2 Signaling Underlies P63 Oncogenic Function in Squamous Cell Carcinoma. J. Clin. Investig. 2013, 123, 3525–3538. [Google Scholar] [CrossRef] [Green Version]
- Saladi, S.V.; Ross, K.; Karaayvaz, M.; Tata, P.R.; Mou, H.; Rajagopal, J.; Ramaswamy, S.; Ellisen, L.W. ACTL6A Is Co-Amplified with P63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell 2017, 31, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Rocco, J.W.; Leong, C.-O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. P63 Mediates Survival in Squamous Cell Carcinoma by Suppression of P73-DepenDent. Apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre-Perona, A.; Hoang-Phou, S.; Leitner, M.-C.; Okuniewska, M.; Meehan, S.; Schober, M. De Novo PITX1 Expression Controls Bi-Stable Transcriptional Circuits to Govern Self-Renewal and Differentiation in Squamous Cell Carcinoma. Cell Stem Cell 2019, 24, 390–404.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef]
- Martin, D.; Degese, M.S.; Vitale-Cross, L.; Iglesias-Bartolome, R.; Valera, J.L.C.; Wang, Z.; Feng, X.; Yeerna, H.; Vadmal, V.; Moroishi, T.; et al. Assembly and Activation of the Hippo Signalome by FAT1 Tumor Suppressor. Nat. Commun. 2018, 9, 2372. [Google Scholar] [CrossRef] [PubMed]
- Namani, A.; Rahaman, M.M.; Chen, M.; Tang, X. Gene-Expression Signature Regulated by the KEAP1-NRF2-CUL3 Axis Is Associated with a Poor Prognosis in Head and Neck Squamous Cell Cancer. BMC Cancer 2018, 18, 46. [Google Scholar] [CrossRef]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Adelstein, D.J.; Moon, J.; Hanna, E.; Giri, P.G.S.; Mills, G.M.; Wolf, G.T.; Urba, S.G. Docetaxel, Cisplatin, and Fluorouracil Induction Chemotherapy Followed by Accelerated Fractionation/Concomitant Boost Radiation and Concurrent Cisplatin in Patients with Advanced Squamous Cell Head and Neck Cancer: A Southwest Oncology Group Phase II Trial (S0216). Head Neck 2010, 32, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus Cetuximab for Locoregionally Advanced Head and Neck Cancer: 5-Year Survival Data from a Phase 3 Randomised Trial, and Relation between Cetuximab-Induced Rash and Survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Gibson, M.K.; Li, Y.; Murphy, B.; Hussain, M.H.A.; DeConti, R.C.; Ensley, J.; Forastiere, A.A.; Group, E.C.O. Randomized Phase III Evaluation of Cisplatin Plus Fluorouracil Versus Cisplatin Plus Paclitaxel in Advanced Head and Neck Cancer (E1395): An Intergroup Trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2005, 23, 3562–3567. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Y.; Hong, S.; Yang, Y.; Yu, G.; Jia, J.; Peng, P.; Wu, X.; Lin, Q.; Xi, X.; et al. Gemcitabine plus Cisplatin versus Fluorouracil plus Cisplatin in Recurrent or Metastatic Nasopharyngeal Carcinoma: A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2016, 388, 1883–1892. [Google Scholar] [CrossRef]
- Jacobs, C.; Lyman, G.; Velez-García, E.; Sridhar, K.S.; Knight, W.; Hochster, H.; Goodnough, L.T.; Mortimer, J.E.; Einhorn, L.H.; Schacter, L. A Phase III Randomized Study Comparing Cisplatin and Fluorouracil as Single Agents and in Combination for Advanced Squamous Cell Carcinoma of the Head and Neck. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1992, 10, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraf, M.; Metch, B.; Kish, J.; Ensley, J.; Rinehart, J.J.; Schuller, D.E.; Coltman, C.A. Platinum Analogs in Recurrent and Advanced Head and Neck Cancer: A Southwest Oncology Group and Wayne State University Study. Cancer Treat Rep. 1987, 71, 723–726. [Google Scholar] [PubMed]
- Catimel, G.; Verweij, J.; Mattijssen, V.; Hanauske, A.; Piccart, M.; Wanders, J.; Franklin, H.; Bail, N.L.; Clavel, M.; Kaye, S.B.; et al. Docetaxel (Taxotere): An Active Drug for the Treatment of Patients with Advanced Squamous Cell Carcinoma of the Head and Neck. EORTC Early Clinical Trials Group. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. ESMO 1994, 5, 533–537. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Trigo, J.; Hitt, R.; Koralewski, P.; Diaz-Rubio, E.; Rolland, F.; Knecht, R.; Amellal, N.; Schueler, A.; Baselga, J. Open-Label, Uncontrolled, Multicenter Phase II Study to Evaluate the Efficacy and Toxicity of Cetuximab as a Single Agent in Patients with Recurrent and/or Metastatic Squamous Cell Carcinoma of the Head and Neck Who Failed to Respond to Platinum-Based Therapy. J. Clin. Oncol. 2007, 25, 2171–2177. [Google Scholar] [CrossRef]
- Chan, A.T.C.; Hsu, M.-M.; Goh, B.C.; Hui, E.P.; Liu, T.-W.; Millward, M.J.; Hong, R.-L.; Whang-Peng, J.; Ma, B.B.Y.; To, K.F.; et al. Multicenter, Phase II Study of Cetuximab in Combination with Carboplatin in Patients with Recurrent or Metastatic Nasopharyngeal Carcinoma. J. Clin. Oncol. 2005, 23, 3568–3576. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; Angelis, F.D.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab Alone or with Chemotherapy versus Cetuximab with Chemotherapy for Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (KEYNOTE-048): A Randomised, Open-Label, Phase 3 Study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Harrington, K.J.; Ferris, R.L.; Blumenschein, G.; Colevas, A.D.; Fayette, J.; Licitra, L.; Kasper, S.; Even, C.; Vokes, E.E.; Worden, F.; et al. Nivolumab versus Standard, Single-Agent Therapy of Investigator’s Choice in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (CheckMate 141): Health-Related Quality-of-Life Results from a Randomised, Phase 3 Trial. Lancet Oncol. 2017, 18, 1104–1115. [Google Scholar] [CrossRef]
- Ho, A.L.; Brana, I.; Haddad, R.; Bauman, J.; Bible, K.; Oosting, S.; Wong, D.J.; Ahn, M.-J.; Boni, V.; Even, C.; et al. Tipifarnib in Head and Neck Squamous Cell Carcinoma with HRAS Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.; Lee, S.-H.; Ejadi, S.; Even, C.; Cohen, R.B.; Tourneau, C.L.; Mehnert, J.M.; Algazi, A.; van Brummelen, E.M.J.; Saraf, S.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients with ProgramMed. Death-Ligand 1–Positive Nasopharyngeal Carcinoma: Results of the KEYNOTE-028 Study. J. Clin. Oncol. 2017, 35, 4050–4056. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; He, J.; Li, B.; Zheng, Y.; Li, K.; Zou, S.; Chen, L. Efficacy and Safety of Gefitinib in Patients with Advanced Head and Neck Squamous Cell Carcinoma: A Meta-Analysis of Randomized Controlled Trials. J. Oncol. 2019, 2019, 6273438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulieres, D.; Senzer, N.N.; Vokes, E.E.; Hidalgo, M.; Agarwala, S.S.; Siu, L.L. Multicenter Phase II Study of Erlotinib, an Oral Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, in Patients with Recurrent or Metastatic Squamous Cell Cancer of the Head and Neck. J. Clin. Oncol. 2003, 22, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Affolter, A.; Samosny, G.; Heimes, A.; Schneider, J.; Weichert, W.; Stenzinger, A.; Sommer, K.; Jensen, A.; Mayer, A.; Brenner, W.; et al. Multikinase Inhibitors Sorafenib and Sunitinib as Radiosensitizers in Head and Neck Cancer Cell Lines. Head Neck 2017, 39, 623–632. [Google Scholar] [CrossRef]
- Machiels, J.H.; Henry, S.; Zanetta, S.; Kaminsky, M.; Michoux, N.; Bompas, E.; Dillies, A.; Faivre, S.; Schmitz, S.; Guigay, J. Phase II Study of Sunitinib in Patients with Recurrent and/or Metastatic Squamous Head and Neck Carcinoma: The GORTEC 2006–01 Study. J. Clin. Oncol. 2009, 27, 6024. [Google Scholar] [CrossRef]
- Kochanny, S.E.; Worden, F.P.; Adkins, D.R.; Lim, D.W.; Bauman, J.E.; Wagner, S.A.; Brisson, R.J.; Karrison, T.G.; Stadler, W.M.; Vokes, E.E.; et al. A Randomized Phase 2 Network Trial of Tivantinib plus Cetuximab versus Cetuximab in Patients with Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma. Cancer 2020, 126, 2146–2152. [Google Scholar] [CrossRef]
- Oppelt, P.; Ley, J.C.; Worden, F.; Palka, K.; Maggiore, R.; Liu, J.; Adkins, D. Palbociclib and Cetuximab in Cetuximab-Resistant Human Papillomavirus-Related Oropharynx Squamous-Cell Carcinoma: A Multicenter Phase 2 Trial. Oral Oncol. 2021, 114, 105164. [Google Scholar] [CrossRef]
- Adkins, D.; Lin, J.-C.; Sacco, A.G.; Ley, J.C.; Oppelt, P.; Shen, Q.; Kern, K.A.; Thurm, H.C.; Wang, S.-L.; Martini, J.-F.; et al. Palbociclib plus Cetuximab versus Placebo plus Cetuximab in Platinum-Resistant, Cetuximab-Naive, HPV-Unrelated Head and Neck Cancer: A Double-Blind Randomized Phase II Trial (PALATINUS). J. Clin. Oncol. 2019, 37, 6013. [Google Scholar] [CrossRef]
- Duffy, E.; Fitzgerald, W.; Boyle, K.; Rohatgi, R. Nephrotoxicity: Evidence in Patients Receiving Cisplatin Therapy. Clin. J. Oncol. Nurse 2018, 22, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eastman, A. The Formation, Isolation and Characterization of DNA Adducts Produced by Anticancer Platinum Complexes. Pharmacol. Ther. 1987, 34, 155–166. [Google Scholar] [CrossRef]
- Blair, B.G.; Larson, C.A.; Adams, P.L.; Abada, P.B.; Pesce, C.E.; Safaei, R.; Howell, S.B. Copper Transporter 2 Regulates Endocytosis and Controls Tumor Growth and Sensitivity to Cisplatin In Vivo. Mol. Pharmacol. 2011, 79, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemer, S.; Fauth, T.; Scholz, P.; Al-Zamel, Y.; Khamis, A.; Gül, D.; Freudelsperger, L.; Wollenberg, B.; Becker, S.; Stauber, R.H.; et al. Profiling Cisplatin Resistance in Head and Neck Cancer: A Critical Role of the VRAC Ion Channel for Chemoresistance. Cancers 2021, 13, 4831. [Google Scholar] [CrossRef]
- Kemp, S.R.M.; Dalm, S.U.; Wijnolts, F.M.J.; Brink, A.; Honeywell, R.J.; Peters, G.J.; Braakhuis, B.J.M.; Brakenhoff, R.H. DNA-Bound Platinum is the Major Determinant of Cisplatin Sensitivity in Head and Neck Squamous Carcinoma Cells. PLoS ONE 2013, 8, e61555. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.-M.; Lin, P.-M.; Chang, J.-G.; Lin, H.-C.; Li, S.-H.; Lin, S.-F.; Yang, M.-Y. Upregulated SLC22A3 Has a Potential for Improving Survival of Patients with Head and Neck Squamous Cell Carcinoma Receiving Cisplatin Treatment. Oncotarget 2017, 8, 74348–74358. [Google Scholar] [CrossRef] [Green Version]
- Theile, D.; Ketabi-Kiyanvash, N.; Herold-Mende, C.; Dyckhoff, G.; Efferth, T.; Bertholet, V.; Haefeli, W.E.; Weiss, J. Evaluation of Drug Transporters' Significance for Multidrug Resistance in Head and Neck Squamous Cell Carcinoma. Head Neck 2011, 33, 959–968. [Google Scholar] [CrossRef]
- Kutler, D.I.; Auerbach, A.D.; Satagopan, J.; Giampietro, P.F.; Batish, S.D.; Huvos, A.G.; Goberdhan, A.; Shah, J.P.; Singh, B. High Incidence of Head and Neck Squamous Cell Carcinoma in Patients with Fanconi Anemia. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Skowron, M.A.; Petzsch, P.; Hardt, K.; Wagner, N.; Beier, M.; Stepanow, S.; Drechsler, M.; Rieder, H.; Köhrer, K.; Niegisch, G.; et al. Distinctive Mutational Spectrum and Karyotype Disruption in Long-Term Cisplatin-Treated Urothelial Carcinoma Cell Lines. Sci. Rep. 2019, 9, 14476. [Google Scholar] [CrossRef]
- Boot, A.; Huang, M.N.; Ng, A.W.T.; Ho, S.-C.; Lim, J.Q.; Kawakami, Y.; Chayama, K.; Teh, B.T.; Nakagawa, H.; Rozen, S.G. In-Depth Characterization of the Cisplatin Mutational Signature in Human Cell Lines and in Esophageal and Liver Tumors. Genome Res. 2018, 28, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- McNeil, E.M.; Melton, D.W. DNA Repair Endonuclease ERCC1–XPF as a Novel Therapeutic Target to Overcome Chemoresistance in Cancer Therapy. Nucleic Acids Res. 2012, 40, 9990–10004. [Google Scholar] [CrossRef] [Green Version]
- Chiu, T.-J.; Chen, C.-H.; Chien, C.-Y.; Li, S.-H.; Tsai, H.-T.; Chen, Y.-J. High ERCC1 Expression Predicts Cisplatin-Based Chemotherapy Resistance and Poor Outcome in Unresectable Squamous Cell Carcinoma of Head and Neck in a Betel-Chewing Area. J. Transl. Med. 2011, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Vaezi, A.; Wang, X.; Buch, S.; Gooding, W.; Wang, L.; Seethala, R.R.; Weaver, D.T.; D’Andrea, A.D.; Argiris, A.; Romkes, M.; et al. XPF Expression Correlates with Clinical Outcome in Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2011, 17, 5513–5522. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous Recombination and the Repair of DNA Double-Strand Breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Burcher, K.M.; Faucheux, A.T.; Lantz, J.W.; Wilson, H.L.; Abreu, A.; Salafian, K.; Patel, M.J.; Song, A.H.; Petro, R.M.; Lycan, T.; et al. Prevalence of DNA Repair Gene Mutations in Blood and Tumor Tissue and Impact on Prognosis and Treatment in HNSCC. Cancers 2021, 13, 3118. [Google Scholar] [CrossRef]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O6-Methylguanine-DNA Methyltransferase with Specific Inhibitors as a Strategy in Cancer Therapy. Cell Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Kuo, C.C.; Li, C.F.; Cheung, C.H.A.; Tsou, T.C.; Chiang, H.C.; Yang, Y.N.; Chang, S.L.; Lin, L.C.; Pan, H.Y.; et al. O6-methylguanine DNA Methyltransferase Repairs Platinum-DNA Adducts Following Cisplatin Treatment and Predicts Prognoses of Nasopharyngeal Carcinoma. Int. J. Cancer 2015, 137, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-C.; Hsiao, J.-R.; Jiang, S.-S.; Chang, J.-Y.; Chu, P.-Y.; Liu, K.-J.; Fang, H.-L.; Lin, L.-M.; Chen, H.-H.; Huang, Y.-W.; et al. C-MYC-Directed NRF2 Drives Malignant Progression of Head and Neck Cancer via Glucose-6-Phosphate Dehydrogenase and Transketolase Activation. Theranostics 2021, 11, 5232–5247. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Jang, H.; Shin, D.; Baek, S.H.; Roh, J.-L. Targeting Nrf2 with Wogonin Overcomes Cisplatin Resistance in Head and Neck Cancer. Apoptosis 2016, 21, 1265–1278. [Google Scholar] [CrossRef]
- Noman, A.S.M.; Parag, R.R.; Rashid, M.I.; Islam, S.; Rahman, M.Z.; Chowdhury, A.A.; Sultana, A.; Jerin, C.; Siddiqua, A.; Rahman, L.; et al. Chemotherapeutic Resistance of Head and Neck Squamous Cell Carcinoma Is Mediated by EpCAM Induction Driven by IL-6/P62 Associated Nrf2-Antioxidant Pathway Activation. Cell Death Dis. 2020, 11, 663. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.-L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of Ferroptotic Cell Death for Overcoming Cisplatin Resistance of Head and Neck Cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; You, J.H.; Shin, D.; Roh, J.-L. Inhibition of Glutaredoxin 5 Predisposes Cisplatin-Resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics 2020, 10, 7775–7786. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.; Blanpain, C. Unravelling Cancer Stem Cell Potential. Nat. Rev. Cancer 2013, 13, 727–738. [Google Scholar] [CrossRef]
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the Cellular Origin of Squamous Skin Tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.T.; Park, C.Y.; Ailles, L.E.; Weissman, I.L. The Cancer Stem Cell Hypothesis: A Work in Progress. Lab. Investig. 2006, 86, 1203–1207. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.B.; Klein, O.D. Oral Epithelial Stem Cells in Tissue Maintenance and Disease: The First Steps in a Long Journey. Int. J. Oral Sci. 2013, 5, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Iglesias-Bartolome, R.; Uchiyama, A.; Molinolo, A.A.; Abusleme, L.; Brooks, S.R.; Callejas-Valera, J.L.; Edwards, D.; Doci, C.; Asselin-Labat, M.-L.; Onaitis, M.W.; et al. Transcriptional Signature Primes Human Oral Mucosa for Rapid Wound Healing. Sci. Transl. Med. 2018, 10, eaap8798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 Is Required for Maintenance of Adult Self-Renewing Haematopoietic Stem Cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Sauvageau, G. Bmi-1 Determines the Proliferative Capacity of Normal and Leukaemic Stem Cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Komai, Y.; Tokuyama, Y.; Yanai, H.; Ohe, S.; Okazaki, K.; Ueno, H. Identification of Stem Cells That Maintain and Regenerate Lingual Keratinized Epithelial Cells. Nat. Cell Biol. 2013, 15, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Atsumi, N.; Nakamura, N.; Yanai, H.; Komai, Y.; Omachi, T.; Tanaka, K.; Ishigaki, K.; Saiga, K.; Ohsugi, H.; et al. Bmi1-Positive Cells in the Lingual Epithelium Could Serve as Cancer Stem Cells in Tongue Cancer. Sci. Rep. 2016, 6, 39386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Wu, M.; Li, Y.; Chang, I.; Yuan, Q.; Ekimyan-Salvo, M.; Deng, P.; Yu, B.; Yu, Y.; Dong, J.; et al. Targeting BMI1+ Cancer Stem Cells Overcomes Chemoresistance and Inhibits Metastases in Squamous Cell Carcinoma. Cell Stem Cell 2017, 20, 621–634.e6. [Google Scholar] [CrossRef] [Green Version]
- Herzog, A.E.; Warner, K.A.; Zhang, Z.; Bellile, E.; Bhagat, M.A.; Castilho, R.M.; Wolf, G.T.; Polverini, P.J.; Pearson, A.T.; Nör, J.E. The IL-6R and Bmi-1 Axis Controls Self-Renewal and Chemoresistance of Head and Neck Cancer Stem Cells. Cell Death Dis. 2021, 12, 988. [Google Scholar] [CrossRef]
- Nör, C.; Zhang, Z.; Warner, K.A.; Bernardi, L.; Visioli, F.; Helman, J.I.; Roesler, R.; Nör, J.E. Cisplatin Induces Bmi-1 and Enhances the Stem Cell Fraction in Head and Neck Cancer. Neoplasia 2014, 16, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-S.; Wu, M.-J.; Huang, C.-Y.; Lin, S.-C.; Chuang, T.-H.; Yu, C.-C.; Lo, J.-F. CD133/Src Axis Mediates Tumor Initiating Property and Epithelial-Mesenchymal Transition of Head and Neck Cancer. PLoS ONE 2011, 6, e28053. [Google Scholar] [CrossRef]
- Lee, J.; Park, M.; Ko, Y.; Kim, B.; Kim, O.; Hyun, H.; Kim, D.; Sohn, H.; Moon, Y.L.; Lim, W. Ectopic Overexpression of CD133 in HNSCC Makes It Resistant to Commonly Used Chemotherapeutics. Tumor Biol. 2017, 39, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhou, J.; Lu, J.; Xiong, H.; Shi, X.; Gong, L. Significance of CD44 Expression in Head and Neck Cancer: A Systemic Review and Meta-Analysis. BMC Cancer 2014, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Wagner, S.; Ma, C.; Coordes, A.; Gekeler, J.; Klussmann, J.P.; Hummel, M.; Kaufmann, A.M.; Albers, A.E. Prognostic Significance of ALDH1A1-Positive Cancer Stem Cells in Patients with Locally Advanced, Metastasized Head and Neck Squamous Cell Carcinoma. J. Cancer Res. Clin. 2014, 140, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Ge, N.N. Cancer Subclones Derived from the Patient’s Head and Neck Squamous Cell Carcinoma Tumor Stem Cells for the Screening of Personalized Antitumor Immunotherapy and Chemotherapy. Stem Cell Res. Ther. 2018, 3, 116–121. [Google Scholar]
- Chen, D.; Wang, C.-Y. Targeting Cancer Stem Cells in Squamous Cell Carcinoma. Precis. Clin. Med. 2019, 2, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem Cell Fate in Cancer Growth, Progression and Therapy Resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour Stem Cells and Drug Resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Chang, C.-W.; Chen, Y.-S.; Chou, S.-H.; Han, C.-L.; Chen, Y.-J.; Yang, C.-C.; Huang, C.-Y.; Lo, J.-F. Distinct Subpopulations of Head and Neck Cancer Cells with Different Levels of Intracellular Reactive Oxygen Species Exhibit Diverse Stemness, Proliferation, and Chemosensitivity. Cancer Res. 2014, 74, 6291–6305. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Yonekubo, Y.; Hanson, N.; Sastre-Perona, A.; Basin, A.; Rytlewski, J.A.; Dolgalev, I.; Meehan, S.; Tsirigos, A.; Beronja, S.; et al. TGF-β-Induced Quiescence Mediates Chemoresistance of Tumor-Propagating Cells in Squamous Cell Carcinoma. Cell Stem Cell 2017, 21, 650–664.e8. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The Metabolic Flexibility of Quiescent CSC: Implications for Chemotherapy Resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Keymeulen, A.V.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the Tumour Transition States Occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial–Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitali, R.; Mancini, C.; Cesi, V.; Tanno, B.; Mancuso, M.; Bossi, G.; Zhang, Y.; Martinez, R.V.; Calabretta, B.; Dominici, C.; et al. Slug (SNAI2) Down-Regulation by RNA Interference Facilitates Apoptosis and Inhibits Invasive Growth in Neuroblastoma Preclinical Models. Clin. Cancer Res. 2008, 14, 4622–4630. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.S.-S.; Lan, H.-Y.; Huang, C.-H.; Tai, S.-K.; Chang, S.-Y.; Tsai, T.-L.; Chang, C.-C.; Tzeng, C.-H.; Wu, K.-J.; Kao, J.-Y.; et al. Regulation of Excision Repair Cross-Complementation Group 1 by Snail Contributes to Cisplatin Resistance in Head and Neck Cancer. Clin. Cancer Res. 2010, 16, 4561–4571. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Z.; Zhang, Q.; Zhang, Q.; Sun, P.; Xiang, R.; Ren, G.; Yang, S. ZEB1 Confers Chemotherapeutic Resistance to Breast Cancer by Activating ATM. Cell Death Dis. 2018, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Gross, K.M.; Zhou, W.; Breindel, J.L.; Ouyang, J.; Jin, D.X.; Sokol, E.S.; Gupta, P.B.; Huber, K.; Zou, L.; Kuperwasser, C. Loss of Slug Compromises DNA Damage Repair and Accelerates Stem Cell Aging in Mammary Epithelium. Cell Rep. 2019, 28, 394–407.e6. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhou, L.; Wei, C.; Zhao, W.; Yu, D. Slug Inhibition Increases Radiosensitivity of Oral Squamous Cell Carcinoma Cells by Upregulating PUMA. Int. J. Oncol. 2016, 49, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Schinke, H.; Pan, M.; Akyol, M.; Zhou, J.; Shi, E.; Kranz, G.; Libl, D.; Quadt, T.; Simon, F.; Canis, M.; et al. SLUG-related Partial Epithelial-to-mesenchymal Transition Is a Transcriptomic Prognosticator of Head and Neck Cancer Survival. Mol. Oncol. 2021, 16, 347–367. [Google Scholar] [CrossRef]
- Sharma, A.; Cao, E.Y.; Kumar, V.; Zhang, X.; Leong, H.S.; Wong, A.M.L.; Ramakrishnan, N.; Hakimullah, M.; Teo, H.M.V.; Chong, F.T.; et al. Longitudinal Single-Cell RNA Sequencing of Patient-Derived Primary Cells Reveals Drug-Induced Infidelity in Stem Cell Hierarchy. Nat. Commun. 2018, 9, 4931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Loo, P.V.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, S.; Larsen, S.B.; Gomez, N.C.; Alaverdyan, K.; Sendoel, A.; Yuan, S.; Polak, L.; Kulukian, A.; Chai, S.; Fuchs, E. Inflammatory Memory Sensitizes Skin Epithelial Stem Cells to Tissue Damage. Nature 2017, 550, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Oh, S.-Y.; Do, S.I.; Lee, H.J.; Kang, H.J.; Rho, Y.S.; Bae, W.J.; Lim, Y.C. SOX2 Regulates Self-Renewal and Tumorigenicity of Stem-like Cells of Head and Neck Squamous Cell Carcinoma. Br. J. Cancer 2014, 111, 2122–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-Function P53 Mutants Co-Opt Chromatin Pathways to Drive Cancer Growth. Nature 2015, 525, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.P.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.-C.; Baslan, T.; Marinkovic, Z.S.; Sánchez-Rivera, F.J.; Leach, S.D.; et al. α-Ketoglutarate Links P53 to Cell Fate during Tumour Suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef]
- Callahan, S.C.; Divenko, M.; Barrodia, P.; Singh, A.K.; Arslan, E.; Liu, Z.; Yang, J.; Anvar, N.; Amit, M.; Xie, T.; et al. KMT2D Loss Promotes Head and Neck Squamous Carcinoma Through Enhancer Reprogramming and Modulation of Immune Microenvironment. bioRxiv 2021. [Google Scholar] [CrossRef]
- Farhangdoost, N.; Horth, C.; Hu, B.; Bareke, E.; Chen, X.; Li, Y.; Coradin, M.; Garcia, B.A.; Lu, C.; Majewski, J. Chromatin Dysregulation Associated with NSD1 Mutation in Head and Neck Squamous Cell Carcinoma. Cell Rep. 2021, 34, 108769. [Google Scholar] [CrossRef]
- Bui, N.; Huang, J.K.; Bojorquez-Gomez, A.; Licon, K.; Sanchez, K.S.; Tang, S.N.; Beckett, A.N.; Wang, T.; Zhang, W.; Shen, J.P.; et al. Disruption of NSD1 in Head and Neck Cancer Promotes Favorable Chemotherapeutic Responses Linked to Hypomethylation. Mol. Cancer Ther. 2018, 17, 1585–1594. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Ji, E.H.; Zhao, X.; Cui, L.; Misuno, K.; Guo, M.; Huang, Z.; Chen, X.; Hu, S. Sox11 Promotes Head and Neck Cancer Progression via the Regulation of SDCCAG8. J. Exp. Clin. Cancer Res. 2019, 38, 138. [Google Scholar] [CrossRef]
- Xie, S.-L.; Fan, S.; Zhang, S.-Y.; Chen, W.-X.; Li, Q.-X.; Pan, G.-K.; Zhang, H.-Q.; Wang, W.-W.; Weng, B.; Zhang, Z.; et al. SOX8 Regulates Cancer Stem-like Properties and Cisplatin-induced EMT in Tongue Squamous Cell Carcinoma by Acting on the Wnt/Β-catenin Pathway. Int. J. Cancer 2018, 142, 1252–1265. [Google Scholar] [CrossRef] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, O.; Jimenez, J.; Pernia, O.; Rodriguez-Antolin, C.; Rodriguez, C.; Cabo, F.S.; Soto, J.; Rosas, R.; Lopez-Magallon, S.; Rodriguez, I.E.; et al. DNA Methylation of MiR-7 Is a Mechanism Involved in Platinum Response through MAFG Overexpression in Cancer Cells. Theranostics 2017, 7, 4118–4134. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Hayward, M.-K.; Weaver, V.M. The Fibrotic Tumor Stroma. Biochim. Biophys. Acta BBA-Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Fiori, M.E.; Franco, S.D.; Villanova, L.; Bianca, P.; Stassi, G.; Maria, R.D. Cancer-Associated Fibroblasts as Abettors of Tumor Progression at the Crossroads of EMT and Therapy Resistance. Mol. Cancer 2019, 18, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, C.; Porter, S. Oral Cancer. West. J. Med. 2001, 174, 348–351. [Google Scholar] [CrossRef]
- Lai, S.L.; Tan, M.L.; Hollows, R.J.; Robinson, M.; Ibrahim, M.; Margielewska, S.; Parkinson, E.K.; Ramanathan, A.; Zain, R.B.; Mehanna, H.; et al. Collagen Induces a More Proliferative, Migratory and Chemoresistant Phenotype in Head and Neck Cancer via DDR1. Cancers 2019, 11, 1766. [Google Scholar] [CrossRef] [Green Version]
- Deville, S.S.; Cordes, N. The Extracellular, Cellular, and Nuclear Stiffness, a Trinity in the Cancer Resistome—A Review. Front. Oncol. 2019, 9, 1376. [Google Scholar] [CrossRef]
- Wang, D.; Müller, S.; Amin, A.R.M.R.; Huang, D.; Su, L.; Hu, Z.; Rahman, M.A.; Nannapaneni, S.; Koenig, L.; Chen, Z.; et al. The Pivotal Role of Integrin Β1 in Metastasis of Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2012, 18, 4589–4599. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.H.; Rho, Y.S.; Lee, S.H.; Koo, B.S.; Lee, H.J.; Do, S.I.; Cho, J.H.; Eun, Y.G.; Park, M.W.; Shin, H.A.; et al. Role of Integrin Β1 as a Biomarker of Stemness in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2019, 96, 34–41. [Google Scholar] [CrossRef]
- Eke, I.; Cordes, N. Focal Adhesion Signaling and Therapy Resistance in Cancer. Semin. Cancer Biol. 2015, 31, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the Extracellular Matrix: Drivers of Tumour Metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- Matuszewska, K.; Pereira, M.; Petrik, D.; Lawler, J.; Petrik, J. Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers 2021, 13, 4444. [Google Scholar] [CrossRef]
- Magnussen, A.L.; Mills, I.G. Vascular Normalisation as the Stepping Stone into Tumour Microenvironment Transformation. Br. J. Cancer 2021, 125, 324–336. [Google Scholar] [CrossRef]
- Ip, C.K.M.; Li, S.-S.; Tang, M.Y.H.; Sy, S.K.H.; Ren, Y.; Shum, H.C.; Wong, A.S.T. Stemness and Chemoresistance in Epithelial Ovarian Carcinoma Cells under Shear Stress. Sci. Rep. 2016, 6, 26788. [Google Scholar] [CrossRef]
- Xu, H.; Zeng, L.; Guan, Y.; Feng, X.; Zhu, Y.; Lu, Y.; Shi, C.; Chen, S.; Xia, J.; Guo, J.; et al. High NEK2 Confers to Poor Prognosis and Contributes to Cisplatin-based Chemotherapy Resistance in Nasopharyngeal Carcinoma. J. Cell Biochem. 2019, 120, 3547–3558. [Google Scholar] [CrossRef] [Green Version]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix Stiffness Induces Epithelial–Mesenchymal Transition and Promotes Chemoresistance in Pancreatic Cancer Cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Smail, M.; Meng, W.; Shyr, Y.; Ye, F.; Fan, K.-H.; Li, X.; Zhou, H.-M.; Bhowmick, N.A. Yes-Associated Protein Expression in Head and Neck Squamous Cell Carcinoma Nodal Metastasis. PLoS ONE 2011, 6, e27529. [Google Scholar] [CrossRef] [Green Version]
- Matte, B.F.; Kumar, A.; Placone, J.K.; Zanella, V.G.; Martins, M.D.; Engler, A.J.; Lamers, M.L. Matrix Stiffness Mechanically Conditions EMT and Migratory Behavior of Oral Squamous Cell Carcinoma. J. Cell Sci. 2018, 132, jcs224360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.J.; Earle, C.; Wong, G.; Bourguignon, L.Y.W. Role of Hyaluronan Synthase 2 to Promote CD44-depenDent. Oral Cavity Squamous Cell Carcinoma Progression. Head Neck 2013, 35, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Z.; Wu, Y.; Wang, Y.; Wang, D.; Zhang, W.; Yuan, H.; Ye, J.; Song, X.; Yang, J.; et al. The Hippo Effector TAZ Promotes Cancer Stemness by Transcriptional Activation of SOX2 in Head Neck Squamous Cell Carcinoma. Cell Death Dis. 2019, 10, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, G.; Domínguez, D.; Elosúa-Bayes, M.; Beckedorff, F.; Laudanna, C.; Bigas, C.; Douillet, D.; Greco, C.; Symeonidi, A.; Hernández, I.; et al. Dietary Palmitic Acid Promotes a Prometastatic Memory via Schwann Cells. Nature 2021, 599, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Shi, H.; Lin, F.; Dasari, S.; Bednash, J.; Thorne, S.; Watkins, S.; Joshi, R.; Thomas, S.M. Enhancement of Head and Neck Squamous Cell Carcinoma Proliferation, Invasion, and Metastasis by Tumor-associated Fibroblasts in Preclinical Models. Head Neck 2014, 36, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Gieniec, K.A.; Butler, L.M.; Worthley, D.L.; Woods, S.L. Cancer-Associated Fibroblasts—Heroes or Villains? Br. J. Cancer 2019, 121, 293–302. [Google Scholar] [CrossRef]
- Han, C.; Liu, T.; Yin, R. Biomarkers for Cancer-Associated Fibroblasts. Biomark. Res. 2020, 8, 64. [Google Scholar] [CrossRef]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Boyd, L.N.C.; Andini, K.D.; Peters, G.J.; Kazemier, G.; Giovannetti, E. Heterogeneity and Plasticity of Cancer-Associated Fibroblasts in the Pancreatic Tumor Microenvironment. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Chen, X.; Song, E. Turning Foes to Friends: Targeting Cancer-Associated Fibroblasts. Nat. Rev. Drug Discov. 2019, 18, 99–115. [Google Scholar] [CrossRef]
- Blise, K.E.; Sivagnanam, S.; Banik, G.L.; Coussens, L.M.; Goecks, J. Single-Cell Spatial Proteomics Analyses of Head and Neck Squamous Cell Carcinoma Reveal Tumor Heterogeneity and Immune ArchitectuRes. Associated with Clinical Outcome. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yavuz, B.G.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer Associated Fibroblasts Sculpt Tumour Microenvironment by Recruiting Monocytes and Inducing Immunosuppressive PD-1+ TAMs. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef] [PubMed]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-Associated Fibroblasts Induce Epithelial–Mesenchymal Transition of Bladder Cancer Cells through Paracrine IL-6 Signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-Associated Fibroblasts Induce Epithelial–Mesenchymal Transition of Breast Cancer Cells through Paracrine TGF-β Signalling. Br. J. Cancer 2013, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, V.; Montoya, C.A.; Donnenberg, V.S.; Sant, S. Interplay between Tumor Microenvironment and Partial EMT as the Driver of Tumor Progression. IScience 2021, 24, 102113. [Google Scholar] [CrossRef]
- Katsuno, Y.; Derynck, R. Epithelial Plasticity, Epithelial-Mesenchymal Transition, and the TGF-β Family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β Promotes Heterogeneity and Drug Resistance in Squamous Cell Carcinoma. Cell 2015, 160, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Yan, M.; Wang, X.; Xu, Q.; Wang, X.; Zhu, X.; Shi, J.; Li, Z.; Zhang, J.; Chen, W. Cancer-Associated Fibroblast-Derived IL-6 Promotes Head and Neck Cancer Progression via the Osteopontin-NF-Kappa B Signaling Pathway. Theranostics 2018, 8, 921–940. [Google Scholar] [CrossRef]
- Patel, A.K.; Vipparthi, K.; Thatikonda, V.; Arun, I.; Bhattacharjee, S.; Sharan, R.; Arun, P.; Singh, S. A Subtype of Cancer-Associated Fibroblasts with Lower Expression of Alpha-Smooth Muscle Actin Suppresses Stemness through BMP4 in Oral Carcinoma. Oncogenesis 2018, 7, 78. [Google Scholar] [CrossRef]
- Costea, D.E.; Hills, A.; Osman, A.H.; Thurlow, J.; Kalna, G.; Huang, X.; Murillo, C.P.; Parajuli, H.; Suliman, S.; Kulasekara, K.K.; et al. Identification of Two Distinct Carcinoma-Associated Fibroblast Subtypes with Differential Tumor-Promoting Abilities in Oral Squamous Cell Carcinoma. Cancer Res. 2013, 73, 3888–3901. [Google Scholar] [CrossRef] [Green Version]
- Peltanova, B.; Liskova, M.; Gumulec, J.; Raudenska, M.; Polanska, H.H.; Vaculovic, T.; Kalfert, D.; Grega, M.; Plzak, J.; Betka, J.; et al. Sensitivity to Cisplatin in Head and Neck Cancer Cells Is Significantly Affected by Patient-Derived Cancer-Associated Fibroblasts. Int. J. Mol. Sci. 2021, 22, 1912. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-Activated PAI-1 Secretion in the Cancer-Associated Fibroblasts with Paracrine Effects Promoting Esophageal Squamous Cell Carcinoma Progression and Causing Chemoresistance. Cell Death Dis. 2018, 9, 759. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Buechler, M.B.; Fu, W.; Turley, S.J. Fibroblast-Macrophage Reciprocal Interactions in Health, Fibrosis, and Cancer. Immunity 2021, 54, 903–915. [Google Scholar] [CrossRef]
- Taniguchi, S.; Elhance, A.; Duzer, A.V.; Kumar, S.; Leitenberger, J.J.; Oshimori, N. Tumor-Initiating Cells Establish an IL-33–TGF-β Niche Signaling Loop to Promote Cancer Progression. Science 2020, 369, eaay1813. [Google Scholar] [CrossRef]
- Wolf, G.T.; Chepeha, D.B.; Bellile, E.; Nguyen, A.; Thomas, D.; McHugh, J.; The University of Michigan Head and Neck SPORE Program. Tumor Infiltrating Lymphocytes (TIL) and Prognosis in Oral Cavity Squamous Carcinoma: A Preliminary Study. Oral Oncol. 2015, 51, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Gomez, K.E.; Wu, F.; Keysar, S.B.; Morton, J.J.; Miller, B.; Chimed, T.-S.; Le, P.N.; Nieto, C.; Chowdhury, F.N.; Tyagi, A.; et al. Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells. Cancer Res. 2020, 80, 4185–4198. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-Associated Fibroblasts Promote an Immunosuppressive Microenvironment through the Induction and Accumulation of Protumoral Macrophages. Oncotarget 2016, 8, 8633–8647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomarat, P.; Banchereau, J.; Davoust, J.; Palucka, A.K. IL-6 Switches the Differentiation of Monocytes from Dendritic Cells to Macrophages. Nat. Immunol. 2000, 1, 510–514. [Google Scholar] [CrossRef]
- Wu, M.-H.; Hong, H.-C.; Hong, T.-M.; Chiang, W.-F.; Jin, Y.-T.; Chen, Y.-L. Targeting Galectin-1 in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis by Downregulating MCP-1/CCL2 Expression. Clin. Cancer Res. 2011, 17, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-DepenDent. Manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maggioni, D.; Pignataro, L.; Garavello, W. T-Helper and T-Regulatory Cells Modulation in Head and Neck Squamous Cell Carcinoma. Oncoimmunology 2017, 6, e1325066. [Google Scholar] [CrossRef] [Green Version]
- Ihara, F.; Sakurai, D.; Horinaka, A.; Makita, Y.; Fujikawa, A.; Sakurai, T.; Yamasaki, K.; Kunii, N.; Motohashi, S.; Nakayama, T.; et al. CD45RA−Foxp3high Regulatory T Cells Have a Negative Impact on the Clinical Outcome of Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Immunother. 2017, 66, 1275–1285. [Google Scholar] [CrossRef]
- Denkert, C.; Loibl, S.; Noske, A.; Roller, M.; Müller, B.M.; Komor, M.; Budczies, J.; Darb-Esfahani, S.; Kronenwett, R.; Hanusch, C.; et al. Tumor-Associated Lymphocytes as an IndepenDent. Predictor of Response to Neoadjuvant Chemotherapy in Breast Cancer. J. Clin. Oncol. 2009, 28, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Watermann, C.; Pasternack, H.; Idel, C.; Ribbat-Idel, J.; Brägelmann, J.; Kuppler, P.; Offermann, A.; Jonigk, D.; Kühnel, M.P.; Schröck, A.; et al. Recurrent HNSCC Harbor an Immunosuppressive Tumor Immune Microenvironment Suggesting Successful Tumor Immune Evasion. Clin. Cancer Res. 2021, 27, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Allen, C.T.; Xiao, R.; Moore, E.; Davis, R.; Park, S.-J.; Spielbauer, K.; Waes, C.V.; Schmitt, N.C. Cisplatin Alters Anti-Tumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol. Res. 2017, 5, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ Functions and Their Regulation at a Glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo Pathway and Human Cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetto, E.; Brenca, M.; Boeri, M.; Verri, C.; Piccinin, E.; Gasparini, P.; Facchinetti, F.; Rossi, S.; Salvatore, G.; Massimino, M.; et al. YAP1 Acts as Oncogenic Target of 11q22 Amplification in Multiple Cancer Subtypes. Oncotarget 2014, 5, 2608–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, E.; Kim, J. The Potential Role of YAP in Head and Neck Squamous Cell Carcinoma. Exp. Mol. Med. 2020, 52, 1264–1274. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in Physiology and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A Signalling Hub of the Tumour Microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechano-Transduction and YAP-DepenDent. Matrix Remodelling Is Required for the Generation and Maintenance of Cancer Associated Fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D.; Huang, K.; Pacal, M.; McCurdy, S.R.; Lu, S.; Aubry, A.; Yu, T.; Wadosky, K.M.; Zhang, L.; Wang, T.; et al. Binary Pan-Cancer Classes with Distinct Vulnerabilities Defined by pro- or Anti-Cancer YAP/TEAD Activity. Cancer Cell 2021, 39, 1115–1134.e12. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Noguchi, K.; Nakano, Y.; Yamamura, M.; Takaoka, K.; Hashimoto-Tamaoki, T.; Kishimoto, H. The Hippo Pathway Transcriptional Co-Activator, YAP, Confers Resistance to Cisplatin in Human Oral Squamous Cell Carcinoma. Int. J. Oncol. 2015, 46, 2364–2370. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, Y.; Zhu, Y.; Yuan, C.; Wang, D.; Zhang, W.; Qi, B.; Qiu, J.; Song, X.; Ye, J.; et al. The Hippo Transducer TAZ Promotes Epithelial to Mesenchymal Transition and Cancer Stem Cell Maintenance in Oral Cancer. Mol. Oncol. 2015, 9, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Yu, X.; Huang, X.; Liu, Z.; Chai, Y.; Yang, L.; Wang, Q.; Li, M.; Zhao, J.; et al. Unbalanced YAP–SOX9 Circuit Drives Stemness and Malignant Progression in Esophageal Squamous Cell Carcinoma. Oncogene 2019, 38, 2042–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-Associated Protein Mediates Immune Reprogramming in Pancreatic Ductal Adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehsanian, R.; Brown, M.; Lu, H.; Yang, X.; Pattatheyil, A.; Yan, B.; Duggal, P.; Chuang, R.; Doondeea, J.; Feller, S.; et al. YAP Dysregulation by Phosphorylation or ΔNp63-Mediated Gene Repression Promotes Proliferation, Survival and Migration in Head and Neck Cancer Subsets. Oncogene 2010, 29, 6160–6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, J.L.; Grandis, J.R.; Bauman, J.E. The STAT3 Pathway as a Therapeutic Target in Head and Neck Cancer: Barriers and Innovations. Oral Oncol. 2016, 56, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaykalova, D.A.; Manola, J.B.; Ozawa, H.; Zizkova, V.; Morton, K.; Bishop, J.A.; Sharma, R.; Zhang, C.; Michailidi, C.; Considine, M.; et al. NF-κB and Stat3 Transcription Factor SignatuRes. Differentiate HPV-positive and HPV-negative Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2015, 137, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Ma, Y.; Zhang, Z.; Zhao, J.; Kobayashi, H.; Zhang, L.; Fu, L. Expression of Stat3 and Notch1 Is Associated with Cisplatin Resistance in Head and Neck Squamous Cell Carcinoma. Oncol. Rep. 2010, 23, 671–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, M.; Pollock, N.I.; Black, J.; DeGrave, K.A.; Wheeler, S.; Freilino, M.L.; Joyce, S.; Lui, V.W.Y.; Zeng, Y.; Chiosea, S.I.; et al. JAK Kinase Inhibition Abrogates STAT3 Activation and Head and Neck Squamous Cell Carcinoma Tumor Growth. Neoplasia 2015, 17, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, W.-M.; Chang, Y.-C.; Yang, Y.-C.; Lin, S.-K.; Chang, P.M.-H.; Hsiao, M. AKR1C1 Controls Cisplatin-Resistance in Head and Neck Squamous Cell Carcinoma through Cross-Talk with the STAT1/3 Signaling Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 245. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Guo, X.; Wang, Y.; Li, X.; Li, H. Rab18 Regulates Proliferation, Invasion and Cisplatin Sensitivity Through STAT3 Signaling in Head and Neck Squamous Cell Carcinoma. Oncotargets Ther. 2020, 13, 4123–4134. [Google Scholar] [CrossRef]
- Fu, L.; Jin, Q.; Dong, Q.; Li, Q. AATF Is Overexpressed in Human Head and Neck Squamous Cell Carcinoma and Regulates STAT3/Survivin Signaling. OncoTargets Ther. 2021, 14, 5237–5248. [Google Scholar] [CrossRef]
- Qu, H.; Jiang, W.; Wang, Y.; Chen, P. STOML2 as a Novel Prognostic Biomarker Modulates Cell Proliferation, Motility and Chemo-Sensitivity via IL6-Stat3 Pathway in Head and Neck Squamous Cell Carcinoma. Am. J. Transl. Res. 2019, 11, 683–695. [Google Scholar]
- Tao, Y.; Shen, H.; Liu, Y.; Li, G.; Huang, Z.; Liu, Y. IL-23R in Laryngeal Cancer: A Cancer Immunoediting Process That Facilitates Tumor Cell Proliferation and Results in Cisplatin Resistance. Carcinogenesis 2021, 42, 118–126. [Google Scholar] [CrossRef]
- Nisar, S.; Yousuf, P.; Masoodi, T.; Wani, N.A.; Hashem, S.; Singh, M.; Sageena, G.; Mishra, D.; Kumar, R.; Haris, M.; et al. Chemokine-Cytokine Networks in the Head and Neck Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4584. [Google Scholar] [CrossRef]
- Man, C.; Lun, S.W.; Hui, J.W.; To, K.; Choy, K.; Chan, A.W.; Chow, C.; Chung, G.T.; Tsao, S.; Yip, T.T.; et al. Inhibition of NOTCH3 Signalling Significantly Enhances Sensitivity to Cisplatin in EBV-associated Nasopharyngeal Carcinoma. J. Pathol. 2012, 226, 471–481. [Google Scholar] [CrossRef]
- Fukusumi, T.; Guo, T.; Sakai, A.; Ando, M.; Ren, S.; Haft, S.; Liu, C.; Amornphimoltham, P.; Gutkind, J.S.; Califano, J. The NOTCH4-HEY1 Pathway Induces Epithelial Mesenchymal Transition in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 24, 619–633. [Google Scholar] [CrossRef] [Green Version]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.R.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory Autophagy in Cancer-Associated Fibroblasts Promotes Head and Neck Cancer Progression and Offers a Novel Therapeutic Target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Mayea, Y.; Mir, C.; Muñoz, L.; Benavente, S.; Castellvi, J.; Temprana, J.; Maggio, V.; Lorente, J.; Paciucci, R.; LLeonart, M.E. Autophagy Inhibition as a Promising Therapeutic Target for Laryngeal Cancer. Carcinogenesis 2019, 40, 1525–1534. [Google Scholar] [CrossRef]
- Wu, J.-S.; Li, L.; Wang, S.-S.; Pang, X.; Wu, J.-B.; Sheng, S.-R.; Tang, Y.-J.; Tang, Y.-L.; Zheng, M.; Liang, X.-H. Autophagy Is Positively Associated with the Accumulation of Myeloid-Derived Suppressor Cells in 4-Nitroquinoline-1-Oxide-Induced Oral Cancer. Oncol. Rep. 2018, 40, 3381–3391. [Google Scholar] [CrossRef]
- Liao, J.; Zhou, B.; Zhuang, X.; Zhuang, P.; Zhang, D.; Chen, W. Cancer-Associated FIbroblasts Confer Cisplatin Resistance of Tongue Cancer via Autophagy Activation. Biomed. Pharmacother. 2018, 97, 1341–1348. [Google Scholar] [CrossRef]





| Approved | Experimental (Ongoing Clinical Trials) |
|---|---|
| First-line treatment in early/advanced stages Surgical excision and Radiotherap Chemotherapy (platin-based (cis/carboplatin), 5-FU, cetuximab) | Small molecules HRASmut Farnesyltransferase inhibitor: Tipifarnib [13] CDK4/6inhibitor: Palbociclib [15] Tyrosine-kinase inhibitors (TKIs):
|
| First-line treatment for recurrent, unresectable and metastatic tumors Chemotherapy Monotherapy:
Nivolumab, Pembrolizumab (anti-PD-1) [28,29,30,31] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griso, A.B.; Acero-Riaguas, L.; Castelo, B.; Cebrián-Carretero, J.L.; Sastre-Perona, A. Mechanisms of Cisplatin Resistance in HPV Negative Head and Neck Squamous Cell Carcinomas. Cells 2022, 11, 561. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030561
Griso AB, Acero-Riaguas L, Castelo B, Cebrián-Carretero JL, Sastre-Perona A. Mechanisms of Cisplatin Resistance in HPV Negative Head and Neck Squamous Cell Carcinomas. Cells. 2022; 11(3):561. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030561
Chicago/Turabian StyleGriso, Ana Belén, Lucía Acero-Riaguas, Beatriz Castelo, José Luis Cebrián-Carretero, and Ana Sastre-Perona. 2022. "Mechanisms of Cisplatin Resistance in HPV Negative Head and Neck Squamous Cell Carcinomas" Cells 11, no. 3: 561. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030561