
EGFR Signaling in Lung Fibrosis


Abstract
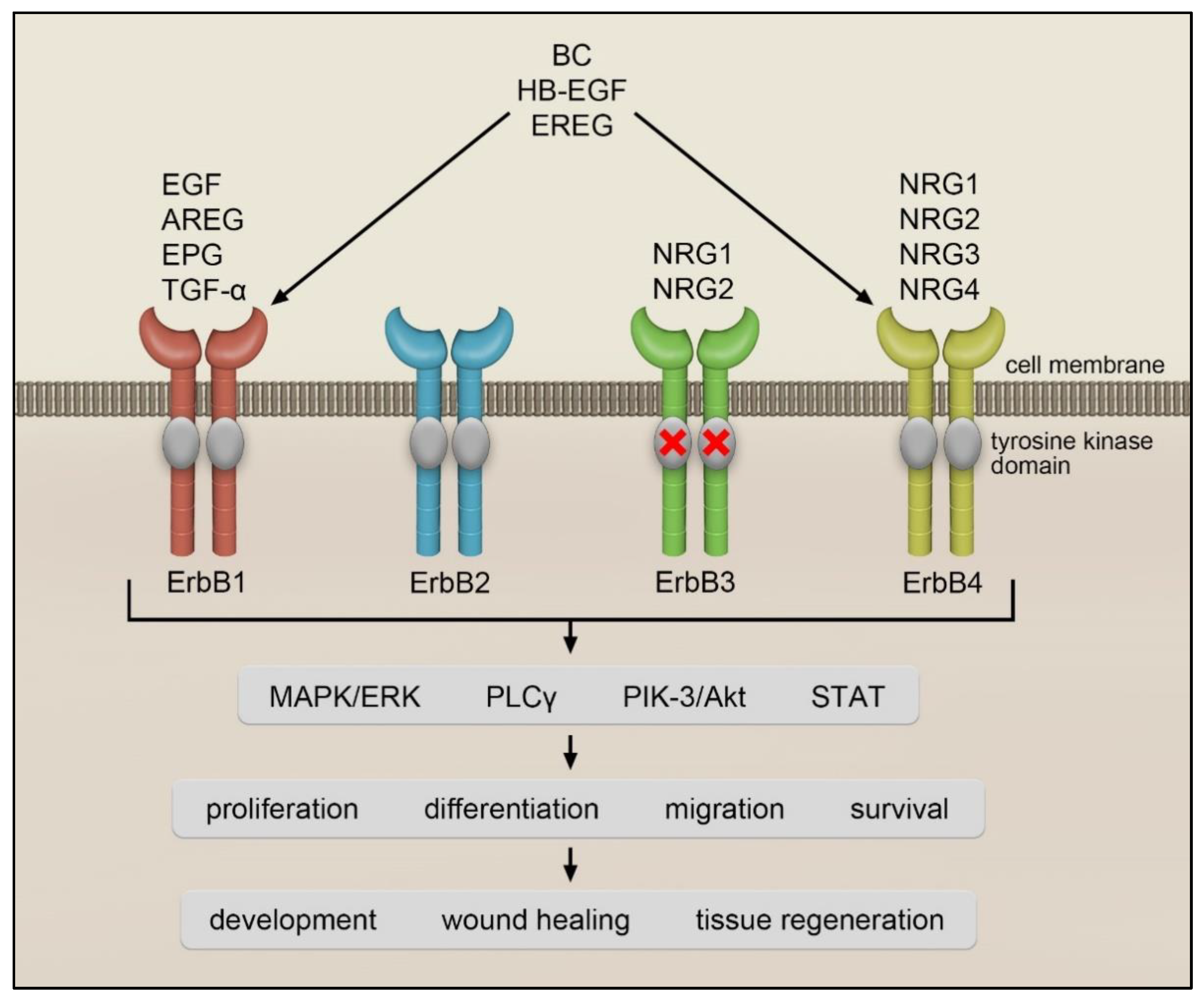
:1. Introduction
2. Idiopathic Pulmonary Fibrosis
3. ErbB Receptor–Ligand System in Lung Fibrosis
3.1. ErbB1/EGFR Receptor
3.2. Transforming Growth Factor-α
3.3. Amphiregulin
3.4. ErbB2 and ErbB3 Receptors and Their Ligands
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, S.; Carpenter, G.; King, L. Epidermal growth factor-receptor-protein kinase interactions. Co-purification of receptor and epidermal growth factor-enhanced phosphorylation activity. J. Biol. Chem. 1980, 255, 4834–4842. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Ikawa, S.; Akiyama, T.; Semba, K.; Nomura, N.; Miyajima, N.; Saito, T.; Toyoshima, K. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature 1986, 319, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.H.; Issing, W.; Miki, T.; Popescu, N.C.; Aaronson, S.A. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: Evidence for overexpression in a subset of human mammary tumors. Proc. Natl. Acad. Sci. USA 1989, 86, 9193–9197. [Google Scholar] [CrossRef] [Green Version]
- Plowman, G.D.; Culouscou, J.M.; Whitney, G.S.; Green, J.M.; Carlton, G.W.; Foy, L.; Neubauer, M.G.; Shoyab, M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl. Acad. Sci. USA 1993, 90, 1746–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S. Isolation of a Mouse Submaxillary Gland Protein Accelerating Incisor Eruption and Eyelid Opening in the New-born Animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar] [CrossRef]
- Jones, S.; Rappoport, J.Z. Interdependent epidermal growth factor receptor signalling and trafficking. Int. J. Biochem. Cell Biol. 2014, 51, 23–28. [Google Scholar] [CrossRef]
- Marquardt, H.; Hunkapiller, M.W.; Hood, L.E.; Todaro, G.J. Rat transforming growth factor type 1: Structure and relation to epidermal growth factor. Science 1984, 223, 1079–1082. [Google Scholar] [CrossRef]
- Shoyab, M.; Plowman, G.D.; McDonald, V.L.; Bradley, J.G.; Todaro, G.J. Structure and function of human amphiregulin: A member of the epidermal growth factor family. Science 1989, 243 Pt 1, 1074–1076. [Google Scholar] [CrossRef]
- Higashiyama, S.; Abraham, J.A.; Miller, J.; Fiddes, J.C.; Klagsbrun, M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science 1991, 251, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Shing, Y.; Christofori, G.; Hanahan, D.; Ono, Y.; Sasada, R.; Igarashi, K.; Folkman, J. Betacellulin: A mitogen from pancreatic beta cell tumors. Science 1993, 259, 1604–1607. [Google Scholar] [CrossRef] [PubMed]
- Komurasaki, T.; Toyoda, H.; Uchida, D.; Morimoto, S. Epiregulin binds to epidermal growth factor receptor and ErbB-4 and induces tyrosine phosphorylation of epidermal growth factor receptor, ErbB-2, ErbB-3 and ErbB-4. Oncogene 1997, 15, 2841–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, L.; Murison, J.G.; Prestidge, R.L.; Sleeman, M.A.; Watson, J.D.; Kumble, K.D. Cloning and biological activity of epigen, a novel member of the epidermal growth factor superfamily. J. Biol. Chem. 2001, 276, 18265–18271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nature reviews. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar]
- Harris, R. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Suzuki, M.; Raab, G.; Moses, M.A.; Fernandez, C.A.; Klagsbrun, M. Matrix metalloproteinase-3 releases active heparin-binding EGF-like growth factor by cleavage at a specific juxtamembrane site. J. Biol. Chem. 1997, 272, 31730–31737. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- NobelPrize.org. Available online: https://www.nobelprize.org/prizes/medicine/1986/summary/ (accessed on 22 January 2022).
- Chang, H.; Riese, D.J.; Gilbert, W.; Stern, D.F.; McMahan, U.J. Ligands for ErbB-family receptors encoded by a neuregulin-like gene. Nature 1997, 387, 509–512. [Google Scholar] [CrossRef]
- Zhang, D.; Sliwkowski, M.X.; Mark, M.; Frantz, G.; Akita, R.; Sun, Y.; Hillan, K.; Crowley, C.; Brush, J.; Godowski, P.J. Neuregulin-3 (NRG3): A novel neural tissue-enriched protein that binds and activates ErbB4. Proc. Natl. Acad. Sci. USA 1997, 94, 9562–9567. [Google Scholar] [CrossRef] [Green Version]
- Harari, D.; Tzahar, E.; Romano, J.; Shelly, M.; Pierce, J.H.; Andrews, G.C.; Yarden, Y. Neuregulin-4: A novel growth factor that acts through the ErbB-4 receptor tyrosine kinase. Oncogene 1999, 18, 2681–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, W.E.; Sliwkowski, M.X.; Akita, R.W.; Henzel, W.J.; Lee, J.; Park, J.W.; Yansura, D.; Abadi, N.; Raab, H.; Lewis, G.D. Identification of heregulin, a specific activator of p185erbB2. Science 1992, 256, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.; Peles, E.; Cupples, R.; Suggs, S.V.; Bacus, S.S.; Luo, Y.; Trail, G.; Hu, S.; Silbiger, S.M.; Levy, R.B.; et al. Neu differentiation factor: A transmembrane glycoprotein containing an EGF domain and an immunoglobulin homology unit. Cell 1992, 69, 559–572. [Google Scholar] [CrossRef]
- Peles, E.; Bacus, S.S.; Koski, R.A.; Lu, H.S.; Wen, D.; Ogden, S.G.; Levy, R.B.; Yarden, Y. Isolation of the NeuHER-2 stimulatory ligand: A 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell 1992, 69, 205–216. [Google Scholar] [CrossRef]
- Falls, D. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell 1993, 72, 801–813. [Google Scholar] [CrossRef]
- Ho, W.H.; Armanini, M.P.; Nuijens, A.; Phillips, H.S.; Osheroff, P.L. Sensory and motor neuron-derived factor. A novel heregulin variant highly expressed in sensory and motor neurons. J. Biol. Chem. 1995, 270, 14523–14532. [Google Scholar] [CrossRef] [Green Version]
- Klapper, L.N.; Glathe, S.; Vaisman, N.; Hynes, N.E.; Andrews, G.C.; Sela, M.; Yarden, Y. The ErbB-2/HER2 oncoprotein of human carcinomas may function solely as a shared coreceptor for multiple stroma-derived growth factors. Proc. Natl. Acad. Sci. USA 1999, 96, 4995–5000. [Google Scholar] [CrossRef] [Green Version]
- Tzahar, E.; Levkowitz, G.; Karunagaran, D.; Yi, L.; Peles, E.; Lavi, S.; Chang, D.; Liu, N.; Yayon, A.; Wen, D. ErbB-3 and ErbB-4 function as the respective low and high affinity receptors of all Neu differentiation factor/heregulin isoforms. J. Biol. Chem. 1994, 269, 25226–25233. [Google Scholar] [CrossRef]
- King, C.R.; Borrello, I.; Bellot, F.; Comoglio, P.; Schlessinger, J. Egf binding to its receptor triggers a rapid tyrosine phosphorylation of the erbB-2 protein in the mammary tumor cell line SK-BR-3. EMBO J. 1988, 7, 1647–1651. [Google Scholar] [CrossRef]
- Sliwkowski, M.X.; Schaefer, G.; Akita, R.W.; Lofgren, J.A.; Fitzpatrick, V.D.; Nuijens, A.; Fendly, B.M.; Cerione, R.A.; Vandlen, R.L.; Carraway, K.L. Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. J. Biol. Chem. 1994, 269, 14661–14665. [Google Scholar] [CrossRef]
- Batzer, A.G.; Rotin, D.; Ureña, J.M.; Skolnik, E.Y.; Schlessinger, J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 1994, 14, 5192–5201. [Google Scholar] [PubMed] [Green Version]
- Rodrigues, G.A.; Falasca, M.; Zhang, Z.; Ong, S.H.; Schlessinger, J. A novel positive feedback loop mediated by the docking protein Gab1 and phosphatidylinositol 3-kinase in epidermal growth factor receptor signaling. Mol. Cell. Biol. 2000, 20, 1448–1459. [Google Scholar] [CrossRef] [Green Version]
- Margolis, B.; Rhee, S.G.; Felder, S.; Mervic, M.; Lyall, R.; Levitzki, A.; Ullrich, A.; Zilberstein, A.; Schlessinger, J. EGF induces tyrosine phosphorylation of phospholipase C-II: A potential mechanism for EGF receptor signaling. Cell 1989, 57, 1101–1107. [Google Scholar] [CrossRef]
- Zeng, F.; Harris, R.C. Epidermal growth factor, from gene organization to bedside. Semin. Cell Dev. Biol. 2014, 28, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Bronson, R.T.; Klaman, L.D.; Hampton, T.G.; Wang, J.F.; Green, P.J.; Magnuson, T.; Douglas, P.S.; Morgan, J.P.; Neel, B.G. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat. Genet. 2000, 24, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Wagner, B.; Hoebertz, A.; Elliott, C.; Marino, S.; Jochum, W.; Wagner, E.F. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 2003, 130, 4515–4525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Luetteke, N.C.; Qiu, T.H.; Peiffer, R.L.; Oliver, P.; Smithies, O.; Lee, D.C. TGFα deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell 1993, 73, 263–278. [Google Scholar] [CrossRef]
- Luetteke, N.C.; Qiu, T.H.; Fenton, S.E.; Troyer, K.L.; Riedel, R.F.; Chang, A.; Lee, D.C. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 1999, 126, 2739–2750. [Google Scholar] [CrossRef]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N. The biology of erbB-2/nue/HER-2 and its role in cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 1994, 1198, 165–184. [Google Scholar] [CrossRef]
- Stancovski, I.; Sela, M.; Yarden, Y. Molecular and clinical aspects of the Neu/ErbB-2 receptor tyrosine kinase. Cancer Treat. Res. 1994, 71, 161–191. [Google Scholar]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.-Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; et al. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) corrected. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Hancock, L.A.; Hennessy, C.E.; Solomon, G.M.; Dobrinskikh, E.; Estrella, A.; Hara, N.; Hill, D.B.; Kissner, W.J.; Markovetz, M.R.; Grove Villalon, D.E.; et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat. Commun. 2018, 9, 5363. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E.; Pardo, A. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Mechanical aspects of lung fibrosis: A spotlight on the myofibroblast. Proc. Am. Thorac. Soc. 2012, 9, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein Shochet, G.; Bardenstein-Wald, B.; Brook, E.; Shitrit, D. Transforming growth factor beta (TGF-β) pathway activation by IPF fibroblast-derived soluble factors is mediated by IL-6 trans-signaling. Eur. Respir. 2020, 56, 3352. [Google Scholar]
- Epstein Shochet, G.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56. [Google Scholar] [CrossRef] [Green Version]
- Kulasekaran, P.; Scavone, C.A.; Rogers, D.S.; Arenberg, D.A.; Thannickal, V.J.; Horowitz, J.C. Endothelin-1 and transforming growth factor-beta1 independently induce fibroblast resistance to apoptosis via AKT activation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.; et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur. Respir. J. 2016, 47, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 1916–1928. [Google Scholar] [CrossRef] [Green Version]
- Kajikawa, K.; Yasui, W.; Sumiyoshi, H.; Yoshida, K.; Nakayama, H.; Ayhan, A.; Yokozaki, H.; Ito, H.; Tahara, E. Expression of epidermal growth factor in human tissues. Immunohistochemical and biochemical analysis. Vichows Arch. A Pathol Anat 1991, 418, 27–32. [Google Scholar] [CrossRef]
- Polosa, R.; Prosperini, G.; Leir, S.H.; Holgate, S.T.; Lackie, P.M.; Davies, D.E. Expression of c-erbB receptors and ligands in human bronchial mucosa. Am. J. Respir. Cell Mol. Biol. 1999, 20, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Aida, S.; Tamai, S.; Sekiguchi, S.; Shimizu, N. Distribution of epidermal growth factor and epidermal growth factor receptor in human lung: Immunohistochemical and immunoelectron-microscopic studies. Respir. Int. Rev. Thorac. Dis. 1994, 61, 161–166. [Google Scholar] [CrossRef]
- Vermeer, P.D.; Panko, L.; Karp, P.; Lee, J.H.; Zabner, J. Differentiation of human airway epithelia is dependent on erbB2. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 291, L175–L180. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Weber, C.K.; Bressau, F.; Greten, F.R.; Stagge, V.; Ebert, M.; Leach, S.D.; Adler, G.; Schmid, R.M. Transgenic overexpression of amphiregulin induces a mitogenic response selectively in pancreatic duct cells. Gastroenterology 2002, 122, 1898–1912. [Google Scholar] [CrossRef]
- Means, A.L.; Ray, K.C.; Singh, A.B.; Washington, M.K.; Whitehead, R.H.; Harris, R.C.; Wright, C.V.E.; Coffey, R.J.; Leach, S.D. Overexpression of heparin-binding EGF-like growth factor in mouse pancreas results in fibrosis and epithelial metaplasia. Gastroenterology 2003, 124, 1020–1036. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Latasa, M.U.; Nicou, A.; Cartagena-Lirola, H.; Castillo, J.; Goñi, S.; Vespasiani-Gentilucci, U.; Zagami, M.G.; Lotersztajn, S.; Prieto, J.; et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008, 48, 1251–1261. [Google Scholar] [CrossRef]
- Dahlhoff, M.; Emrich, D.; Wolf, E.; Schneider, M.R. Increased activation of the epidermal growth factor receptor in transgenic mice overexpressing epigen causes peripheral neuropathy. Biochim. Biophys. Acta 2013, 1832, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Takemura, T.; Yoshida, Y.; Kiso, S.; Kizu, T.; Furuta, K.; Ezaki, H.; Hamano, M.; Egawa, M.; Chatani, N.; Kamada, Y.; et al. Conditional loss of heparin-binding EGF-like growth factor results in enhanced liver fibrosis after bile duct ligation in mice. Biochem. Biophys. Res. Commun. 2013, 437, 185–191. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Ntolios, P.; Karameris, A.; Vilaras, G.; Boglou, P.; Koulelidis, A.; Archontogeorgis, K.; Kaltsas, K.; Zacharis, G.; Sarikloglou, E.; et al. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. BioMed Res. Int. 2013, 2013, 654354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein Shochet, G.; Brook, E.; Eyal, O.; Edelstein, E.; Shitrit, D. Epidermal growth factor receptor paracrine upregulation in idiopathic pulmonary fibrosis fibroblasts is blocked by nintedanib. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L1025–L1034. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.B.; Moomaw, C.R.; Morgan, D.L.; Bonner, J.C. Specific Inhibitors of Platelet-Derived Growth Factor or Epidermal Growth Factor Receptor Tyrosine Kinase Reduce Pulmonary Fibrosis in Rats. Am. J. Pathol. 1999, 155, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Tian, Q.; Liang, Z.; Yang, Z.; Xu, S.; Sun, J.; Chen, L. Gefitinib attenuates murine pulmonary fibrosis induced by bleomycin. Chin. Med. J. 2010, 123, 2259–2264. [Google Scholar] [PubMed]
- Ishii, Y.; Fujimoto, S.; Fukuda, T. Gefitinib prevents bleomycin-induced lung fibrosis in mice. Am. J. Respir. Crit. Care Med. 2006, 174, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Aoshiba, K.; Yokohori, N.; Nagai, A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res. 2003, 63, 5054–5059. [Google Scholar] [PubMed]
- Inomata, S.; Takahashi, H.; Nagata, M.; Yamada, G.; Shiratori, M.; Tanaka, H.; Satoh, M.; Saitoh, T.; Sato, T.; Abe, S. Acute lung injury as an adverse event of gefitinib. Anti-Cancer Drugs 2004, 15, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Saijo, Y.; Maemondo, M.; Gomi, K.; Tokue, Y.; Kimura, Y.; Ebina, M.; Kikuchi, T.; Moriya, T.; Nukiwa, T. Severe acute interstitial pneumonia and gefitinib. Lancet 2003, 361, 137–139. [Google Scholar] [CrossRef]
- Ten Heine, R.; van den Bosch, R.T.A.; Schaefer-Prokop, C.M.; Lankheet, N.A.G.; Beijnen, J.H.; Staaks, G.H.A.; van der Westerlaken, M.M.; Malingré, M.M.; van den Brand, J.J.G. Fatal interstitial lung disease associated with high erlotinib and metabolite levels. A case report and a review of the literature. Lung Cancer 2012, 75, 391–397. [Google Scholar] [CrossRef]
- Ren, S.; Li, Y.; Li, W.; Zhao, Z.; Jin, C.; Zhang, D. Fatal asymmetric interstitial lung disease after erlotinib for lung cancer. Respir. Int. Rev. Thorac. Dis. 2012, 84, 431–435. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA drug approval summary: Gefitinib (ZD1839) (Iressa) tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Inoue, A.; Mitsudomi, T.; Kobayashi, K.; Nakagawa, K.; Mori, K.; Nukiwa, T.; Nakanishi, Y.; Yamamoto, N. Interstitial lung disease associated with gefitinib in Japanese patients with EGFR-mutated non-small-cell lung cancer: Combined analysis of two Phase III trials (NEJ 002 and WJTOG 3405). Jpn. J. Clin. Oncol. 2013, 43, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Okamoto, I.; Yamamoto, N.; Takeda, K.; Tamura, K.; Seto, T.; Ariyoshi, Y.; Fukuoka, M. Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 2549–2556. [Google Scholar] [CrossRef]
- Hardie, W.D.; Davidson, C.; Ikegami, M.; Leikauf, G.D.; Le Cras, T.D.; Prestridge, A.; Whitsett, J.A.; Korfhagen, T.R. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2008, 294, L1217–L1725. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wei, R.; Jones-Hall, Y.L.; Vittal, R.; Zhang, M.; Liu, W. Epidermal growth factor receptor (EGFR) pathway genes and interstitial lung disease: An association study. Sci. Rep. 2014, 4, 4893. [Google Scholar] [CrossRef] [Green Version]
- Baughman, R.P.; Lower, E.E.; Miller, M.A.; Bejarano, P.A.; Heffelfinger, S.C. Overexpression of transforming growth factor-alpha and epidermal growth factor-receptor in idiopathic pulmonary fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. Off. J. WASOG 1999, 16, 57–61. [Google Scholar]
- Madtes, D.K.; Rubenfeld, G.; Klima, L.D.; Milberg, J.A.; Steinberg, K.P.; Martin, T.R.; Raghu, G.; Hudson, L.D.; Clark, J.G. Elevated transforming growth factor-alpha levels in bronchoalveolar lavage fluid of patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1998, 158, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, W.D.; Le Cras, T.D.; Jiang, K.; Tichelaar, J.W.; Azhar, M.; Korfhagen, T.R. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2004, 286, L741–L749. [Google Scholar] [CrossRef] [PubMed]
- Madtes, D.K.; Busby, H.K.; Strandjord, T.P.; Clark, J.G. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am. J. Respir. Cell Mol. Biol. 1994, 11, 540–551. [Google Scholar] [CrossRef]
- Madtes, D.K.; Elston, A.L.; Hackman, R.C.; Dunn, A.R.; Clark, J.G. Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am. J. Respir. Cell Mol. Biol. 1999, 20, 924–934. [Google Scholar] [CrossRef]
- Hardie, W.D.; Korfhagen, T.R.; Sartor, M.A.; Prestridge, A.; Medvedovic, M.; Le Cras, T.D.; Ikegami, M.; Wesselkamper, S.C.; Davidson, C.; Dietsch, M.; et al. Genomic profile of matrix and vasculature remodeling in TGF-alpha induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2007, 37, 309–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, W.D.; Hagood, J.S.; Dave, V.; Perl, A.-K.T.; Whitsett, J.A.; Korfhagen, T.R.; Glasser, S. Signaling pathways in the epithelial origins of pulmonary fibrosis. Cell Cycle 2010, 9, 2769–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madala, S.K.; Schmidt, S.; Davidson, C.; Ikegami, M.; Wert, S.; Hardie, W.D. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am. J. Respir. Cell Mol. Biol. 2012, 46, 380–388. [Google Scholar] [CrossRef]
- Zhou, Y.; Lee, J.-Y.; Lee, C.-M.; Cho, W.-K.; Kang, M.-J.; Koff, J.L.; Yoon, P.-O.; Chae, J.; Park, H.-O.; Elias, J.A.; et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J. Biol. Chem. 2012, 287, 41991–42000. [Google Scholar] [CrossRef] [Green Version]
- Andrianifahanana, M.; Wilkes, M.C.; Repellin, C.E.; Edens, M.; Kottom, T.J.; Rahimi, R.A.; Leof, E.B. ERBB receptor activation is required for profibrotic responses to transforming growth factor beta. Cancer Res. 2010, 70, 7421–7430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrianifahanana, M.; Wilkes, M.C.; Gupta, S.K.; Rahimi, R.A.; Repellin, C.E.; Edens, M.; Wittenberger, J.; Yin, X.; Maidl, E.; Becker, J.; et al. Profibrotic TGFβ responses require the cooperative action of PDGF and ErbB receptor tyrosine kinases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4444–4454. [Google Scholar] [CrossRef] [Green Version]
- Mongera, A.; Rowghanian, P.; Gustafson, H.J.; Shelton, E.; Kealhofer, D.A.; Carn, E.K.; Serwane, F.; Lucio, A.A.; Giammona, J.; Campàs, O. A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature 2018, 561, 401–405. [Google Scholar] [CrossRef]
- Atia, L.; Fredberg, J.J.; Gov, N.S.; Pegoraro, A.F. Are cell jamming and unjamming essential in tissue development? Cells Dev. 2021, 4, 203727. [Google Scholar] [CrossRef]
- Bi, D.; Lopez, J.H.; Schwarz, J.M.; Manning, M.L. A density-independent glass transition in biological tissues. Nat. Phys. 2015, 11, 1074–1079. [Google Scholar] [CrossRef] [Green Version]
- Palamidessi, A.; Malinverno, C.; Frittoli, E.; Corallino, S.; Barbieri, E.; Sigismund, S.; Beznoussenko, G.V.; Martini, E.; Garre, M.; Ferrara, I.; et al. Unjamming overcomes kinetic and proliferation arrest in terminally differentiated cells and promotes collective motility of carcinoma. Nat. Mater. 2019, 18, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-A.; Atia, L.; Mitchel, J.A.; Fredberg, J.J.; Butler, J.P. Collective migration and cell jamming in asthma, cancer and development. J. Cell Sci. 2016, 129, 3375–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stancil, I.T.; Michalski, J.E.; Davis-Hall, D.; Chu, H.W.; Park, J.-A.; Magin, C.M.; Yang, I.V.; Smith, B.J.; Dobrinskikh, E.; Schwartz, D.A. Pulmonary fibrosis distal airway epithelia are dynamically and structurally dysfunctional. Nat. Commun. 2021, 12, 4566. [Google Scholar] [CrossRef] [PubMed]
- Lange, A.W.; Sridharan, A.; Xu, Y.; Stripp, B.R.; Perl, A.-K.; Whitsett, J.A. Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J. Mol. Cell Biol. 2015, 7, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-Y.; Lee, H.; Kwon, E.-J.; Kong, H.-J.; Kwon, O.-S.; Cha, H.-J. TGFβ promotes YAP-dependent AXL induction in mesenchymal-type lung cancer cells. Mol. Oncol. 2021, 15, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Minutti, C.M.; Modak, R.V.; Macdonald, F.; Li, F.; Smyth, D.J.; Dorward, D.A.; Blair, N.; Husovsky, C.; Muir, A.; Giampazolias, E.; et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 2019, 50, 645–654.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukumoto, J.; Harada, C.; Kawaguchi, T.; Suetsugu, S.; Maeyama, T.; Inoshima, I.; Hamada, N.; Kuwano, K.; Nakanishi, Y. Amphiregulin attenuates bleomycin-induced pneumopathy in mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2010, 298, L131–L138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeer, P.D.; Einwalter, L.A.; Moninger, T.O.; Rokhlina, T.; Kern, J.A.; Zabner, J.; Welsh, M.J. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 2003, 422, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Nethery, D.E.; Moore, B.B.; Minowada, G.; Carroll, J.; Faress, J.A.; Kern, J.A. Expression of mutant human epidermal receptor 3 attenuates lung fibrosis and improves survival in mice. J. Appl. Physiol. 2005, 99, 298–307. [Google Scholar] [CrossRef]
- Faress, J.A.; Nethery, D.E.; Kern, E.F.O.; Eisenberg, R.; Jacono, F.J.; Allen, C.L.; Kern, J.A. Bleomycin-induced pulmonary fibrosis is attenuated by a monoclonal antibody targeting HER2. J. Appl. Physiol. 2007, 103, 2077–2083. [Google Scholar] [CrossRef] [Green Version]
- Plantier, L.; Crestani, B.; Wert, S.E.; Dehoux, M.; Zweytick, B.; Guenther, A.; Whitsett, J.A. Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 2011, 66, 651–657. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettle, R.; Simmons, J.; Schindler, F.; Jones, P.; Dicker, T.; Dubois, G.; Giddings, J.; van Heeke, G.; Jones, C.E. Regulation of neuregulin 1beta1-induced MUC5AC and MUC5B expression in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 2010, 42, 472–481. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Targeted Molecule | Inhibitor/ Antibody | Animal Model | Outcome | Reference |
|---|---|---|---|---|
| ErbB1 | AG1478 | Vanadium pentoxide | Favorable | Rice et al., 1999 [73] |
| ErbB1 | Gefitinib | Bleomycin | Favorable | Ishii et al., 2006 [75]; Wang et al., 2010 [74] |
| ErbB1 | Gefitinib | Bleomycin | Harmful | Suzuki et al., 2003 [76] |
| ErbB1 | Gefitinib | rtTA-CCSP 4-TGF-α 5 | Favorable | Hardie et al. 2004 [88] |
| PI3K 1 | PX-866 | rtTA-CCSP-TGF-α | Favorable | Hardie et al., 2010 [92] |
| MEK 2 | ARRY-142886 | rtTA-CCSP-TGF-α | Favorable | Madala et al., 2012 [93] |
| AREG 3 | AREG siRNA | dox-CC10 6-TGF-β1 7 | Favorable | Zhou et al., 2012 [94] |
| ErbB1 | AG1478 | dox-CC10-TGF-β1 | Favorable | Zhou et al., 2012 [94] |
| ErbB1/2 | Lapatinib | Bleomycin | Favorable | Andrianifahanana et al., 2013 [96] |
| ErbB2 | anti-ErbB2 antibody (2C4) | Bleomycin | Favorable | Faress et al., 2007 [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schramm, F.; Schaefer, L.; Wygrecka, M. EGFR Signaling in Lung Fibrosis. Cells 2022, 11, 986. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11060986
Schramm F, Schaefer L, Wygrecka M. EGFR Signaling in Lung Fibrosis. Cells. 2022; 11(6):986. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11060986
Chicago/Turabian StyleSchramm, Fabian, Liliana Schaefer, and Malgorzata Wygrecka. 2022. "EGFR Signaling in Lung Fibrosis" Cells 11, no. 6: 986. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11060986