
Mass Spectrometry for Neurobiomarker Discovery: The Relevance of Post-Translational Modifications

 ,
,  , ,
, , 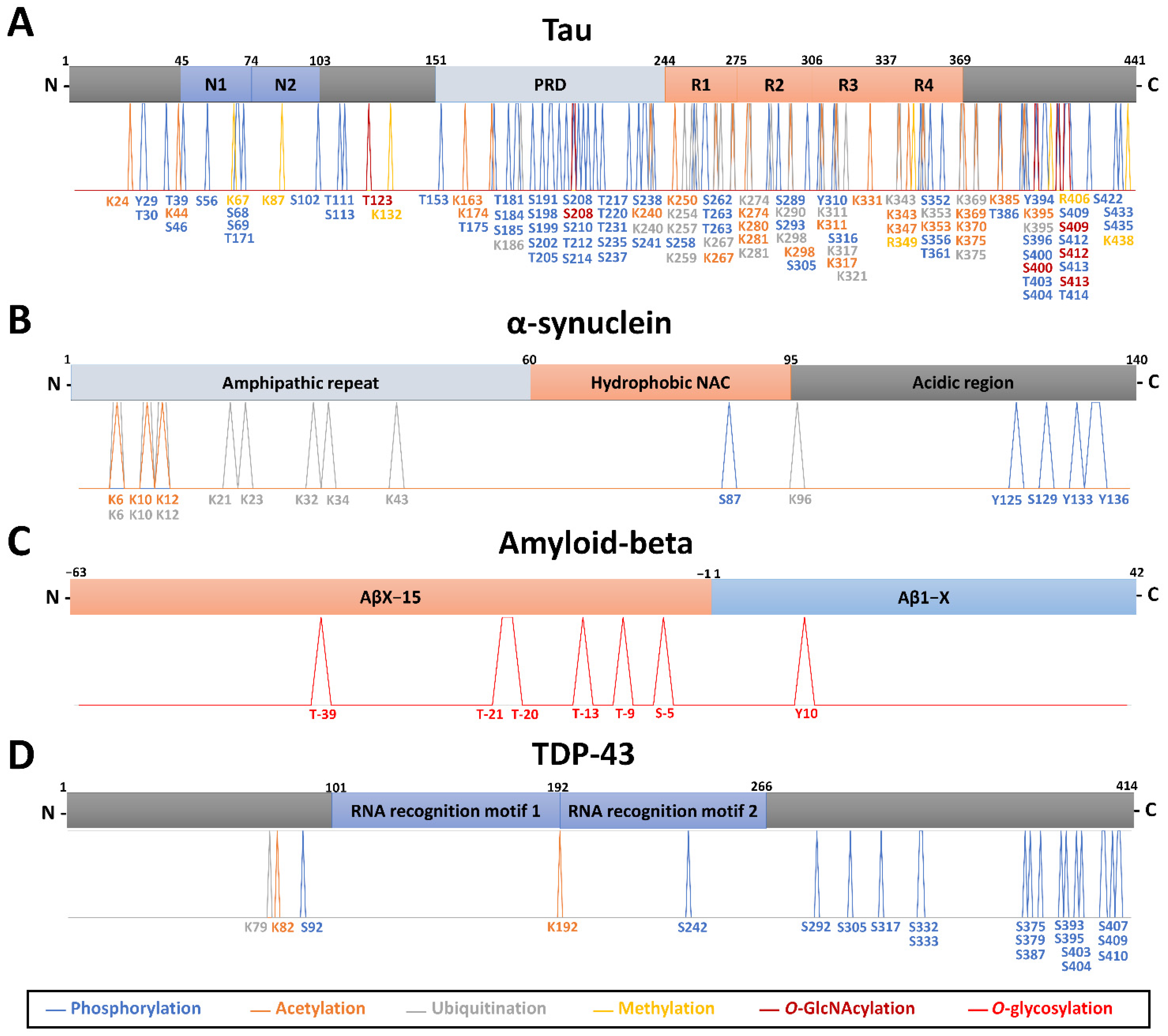{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mass Spectrometry for Neurobiomarker Discovery
2.1. Proteomics with Mass Spectrometry
2.2. Determination of Post-Translational Modifications Using Mass Spectrometry
2.3. PTM-Focused Neuroproteomics: Relevance and Challenges
3. PTM-Proteomic Profiling in Neurodegenerative Diseases by MS
3.1. High-Throughput Profiling of Brain PTMs
3.1.1. Phosphorylation
3.1.2. Ubiquitination
3.1.3. Glycosylation
3.2. Profiling the Main Players in Neurodegeneration
3.2.1. Tau Protein
3.2.2. α-Synuclein
3.2.3. Amyloid Precursor Protein (APP) and Amyloid-Beta Peptides
3.2.4. TAR DNA-Binding Protein (TDP-43)
4. The Next Step in Neuroproteomics
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nichols, E.; Steinmetz, J.D.; Vollset, S.E.; Fukutaki, K.; Chalek, J.; Abd-Allah, F.; Abdoli, A.; Abualhasan, A.; Abu-Gharbieh, E.; Akram, T.T.; et al. Estimation of the Global Prevalence of Dementia in 2019 and Forecasted Prevalence in 2050: An Analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Armstrong, R. What Causes Neurodegenerative Disease? Folia Neuropathol. 2020, 58, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The Probabilistic Model of Alzheimer Disease: The Amyloid Hypothesis Revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Poddar, M.K.; Chakraborty, A.; Banerjee, S. Neurodegeneration: Diagnosis, Prevention, and Therapy; IntechOpen: London, UK, 2021; ISBN 978-1-83880-901-0. [Google Scholar]
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical Diagnosis of Alzheimer’s Disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing Research Diagnostic Criteria for Alzheimer’s Disease: The IWG-2 Criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Oudart, J.-B.; Djerada, Z.; Nonnonhou, V.; Badr, S.; Bertholon, L.-A.; Dammak, A.; Jaidi, Y.; Novella, J.-L.; Pallet, N.; Gillery, P.; et al. Incremental Value of CSF Biomarkers in Clinically Diagnosed AD and Non-AD Dementia. Front. Neurol. 2020, 11, 560. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and Blood Biomarkers for the Diagnosis of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Abeliovich, A.; Gitler, A.D. Defects in Trafficking Bridge Parkinson’s Disease Pathology and Genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef]
- Canter, R.G.; Penney, J.; Tsai, L.-H. The Road to Restoring Neural Circuits for the Treatment of Alzheimer’s Disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T. Ageing, Neurodegeneration and Brain Rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Heemels, M.-T. Neurodegenerative Diseases. Nature 2016, 539, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, D.C.; Brown, J.A.; Possin, K.L.; Datta, S.; Trujillo, A.; Radke, A.; Karydas, A.; Kornak, J.; Sias, A.C.; Rabinovici, G.D.; et al. Clinicopathological Correlations in Behavioural Variant Frontotemporal Dementia. Brain 2017, 140, 3329–3345. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Lee, E.B.; Xie, S.X.; Rennert, L.; Suh, E.; Bredenberg, C.; Caswell, C.; Van Deerlin, V.M.; Yan, N.; Yousef, A.; et al. Neurodegenerative Disease Concomitant Proteinopathies Are Prevalent, Age-Related and APOE4-Associated. Brain 2018, 141, 2181–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrenberg, A.J.; Khatun, A.; Coomans, E.; Betts, M.J.; Capraro, F.; Thijssen, E.H.; Senkevich, K.; Bharucha, T.; Jafarpour, M.; Young, P.N.E.; et al. Relevance of Biomarkers across Different Neurodegenerative. Alzheimers Res. 2020, 12, 56. [Google Scholar] [CrossRef]
- Marsh, A.P. Molecular Mechanisms of Proteinopathies across Neurodegenerative Disease: A Review. Neurol. Res. Pract. 2019, 1, 35. [Google Scholar] [CrossRef] [Green Version]
- Dovidchenko, N.V.; Leonova, E.I.; Galzitskaya, O.V. Mechanisms of Amyloid Fibril Formation. Biochemistry 2014, 79, 1515–1527. [Google Scholar] [CrossRef]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer’s Disease (AD). Int. J. Mol. Sci. 2019, 20, E4661. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Bu, L.-L.; Huang, K.-X.; Zheng, D.-Z.; Lin, D.-Y.; Chen, Y.; Jing, X.-N.; Liang, Y.-R.; Tao, E.-X. Alpha-Synuclein Accumulation and Its Phosphorylation in the Enteric Nervous System of Patients Without Neurodegeneration: An Explorative Study. Front. Aging Neurosci. 2020, 12, 575481. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O. Biomarkers for Neurodegenerative Diseases. Nat. Med. 2021, 27, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Bateman, R.J.; Hirtz, C.; Marin, P.; Becher, F.; Sato, C.; Gabelle, A.; Lehmann, S. Cerebrospinal Fluid Phospho-Tau T217 Outperforms T181 as a Biomarker for the Differential Diagnosis of Alzheimer’s Disease and PET Amyloid-Positive Patient Identification. Alzheimers Res. 2020, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrocki, P.; Khatun, A.; Ness, D.; Senkevich, K.; Hanrieder, J.; Capraro, F.; Mattsson, N.; Andreasson, U.; Portelius, E.; Ashton, N.J.; et al. Perspectives in Fluid Biomarkers in Neurodegeneration from the 2019 Biomarkers in Neurodegenerative Diseases Course-a Joint PhD Student Course at University College London and University of Gothenburg. Alzheimers Res. 2020, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Kelleher, N.L. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.; Latypova, X.; Terro, F. Post-Translational Modifications of Tau Protein: Implications for Alzheimer’s Disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef]
- Olzscha, H. Posttranslational Modifications and Proteinopathies: How Guardians of the Proteome Are Defeated. Biol. Chem. 2019, 400, 895–915. [Google Scholar] [CrossRef]
- Ramesh, M.; Gopinath, P.; Govindaraju, T. Role of Post-Translational Modifications in Alzheimer’s Disease. Chembiochem 2020, 21, 1052–1079. [Google Scholar] [CrossRef]
- Ping, L.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global Quantitative Analysis of the Human Brain Proteome in Alzheimer’s and Parkinson’s Disease. Sci. Data 2018, 5, 180036. [Google Scholar] [CrossRef] [Green Version]
- Ping, L.; Kundinger, S.R.; Duong, D.M.; Yin, L.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Global Quantitative Analysis of the Human Brain Proteome and Phosphoproteome in Alzheimer’s Disease. Sci. Data 2020, 7, 315. [Google Scholar] [CrossRef]
- Abreha, M.H.; Dammer, E.B.; Ping, L.; Zhang, T.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. Quantitative Analysis of the Brain Ubiquitylome in Alzheimer’s Disease. Proteomics 2018, 18, e1800108. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Huo, Z.; Yang, J.; Palma-Gudiel, H.; Boyle, P.A.; Schneider, J.A.; Bennett, D.A.; Zhao, J. Human Brain and Blood N-Glycome Profiling in Alzheimer’s Disease and Alzheimer’s Disease-Related Dementias. Front. Aging Neurosci. 2021, 13, 765259. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Pires, G.; MacMurray, C.; Askenazi, M.; Nayak, S.; Bourdon, M.; Safar, J.; Ueberheide, B.; Wisniewski, T. Phosphorylated Tau Interactome in the Human Alzheimer’s Disease Brain. Brain 2020, 143, 2803–2817. [Google Scholar] [CrossRef]
- Crutchfield, C.A.; Thomas, S.N.; Sokoll, L.J.; Chan, D.W. Advances in Mass Spectrometry-Based Clinical Biomarker Discovery. Clin. Proteom. 2016, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabet, J.-C.; Rebuffat, S. Nobel Prize 2002 for chemistry: Mass spectrometry and nuclear magnetic resonance. Med. Sci. (Paris) 2003, 19, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongré, A.R.; Eng, J.K.; Yates, J.R. Emerging Tandem-Mass-Spectrometry Techniques for the Rapid Identification of Proteins. Trends Biotechnol. 1997, 15, 418–425. [Google Scholar] [CrossRef]
- Marouga, R.; David, S.; Hawkins, E. The Development of the DIGE System: 2D Fluorescence Difference Gel Analysis Technology. Anal. Bioanal Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef]
- Silva, A.M.N.; Vitorino, R.; Domingues, M.R.M.; Spickett, C.M.; Domingues, P. Post-Translational Modifications and Mass Spectrometry Detection. Free Radic. Biol. Med. 2013, 65, 925–941. [Google Scholar] [CrossRef] [Green Version]
- Sajic, T.; Liu, Y.; Aebersold, R. Using Data-Independent, High-Resolution Mass Spectrometry in Protein Biomarker Research: Perspectives and Clinical Applications. Proteom. Clin. Appl. 2015, 9, 307–321. [Google Scholar] [CrossRef]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [Green Version]
- Ke, M.; Shen, H.; Wang, L.; Luo, S.; Lin, L.; Yang, J.; Tian, R. Identification, Quantification, and Site Localization of Protein Posttranslational Modifications via Mass Spectrometry-Based Proteomics. Adv. Exp. Med. Biol. 2016, 919, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, G.; Konzer, A.; Musunuri, S.; Bergquist, J. Neuroproteomics Tools in Clinical Practice. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2015, 1854, 705–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative Mass Spectrometry in Proteomics: A Critical Review. Anal. Bioanal Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics*. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, B.; Tan, H.; Pagala, V.R.; High, A.A.; Ichhaporia, V.P.; Hendershot, L.; Peng, J. Deep Profiling of Proteome and Phosphoproteome by Isobaric Labeling, Extensive Liquid Chromatography and Mass Spectrometry. Methods Enzym. 2017, 585, 377–395. [Google Scholar] [CrossRef] [Green Version]
- Larance, M.; Lamond, A.I. Multidimensional Proteomics for Cell Biology. Nat. Rev. Mol. Cell Biol. 2015, 16, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Rayaprolu, S.; Higginbotham, L.; Bagchi, P.; Watson, C.M.; Zhang, T.; Levey, A.I.; Rangaraju, S.; Seyfried, N.T. Systems-Based Proteomics to Resolve the Biology of Alzheimer’s Disease beyond Amyloid and Tau. Neuropsychopharmacology 2021, 46, 98–115. [Google Scholar] [CrossRef]
- Choudhary, C.; Mann, M. Decoding Signalling Networks by Mass Spectrometry-Based Proteomics. Nat. Rev. Mol. Cell Biol. 2010, 11, 427–439. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-Translational Modifications in Proteins: Resources, Tools and Prediction Methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Fert-Bober, J.; Murray, C.I.; Parker, S.J.; Van Eyk, J.E. Precision Profiling of the Cardiovascular Post-Translationally Modified Proteome: Where There Is a Will, There Is a Way. Circ. Res. 2018, 122, 1221–1237. [Google Scholar] [CrossRef]
- Mann, M.; Jensen, O.N. Proteomic Analysis of Post-Translational Modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Pagel, O.; Loroch, S.; Sickmann, A.; Zahedi, R. Current Strategies and Findings in Clinically Relevant Post-Translational Modification-Specific Proteomics. Expert Rev. Proteom. 2015, 12, 235–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leutert, M.; Entwisle, S.W.; Villén, J. Decoding Post-Translational Modification Crosstalk With Proteomics. Mol. Cell Proteom. 2021, 20, 100129. [Google Scholar] [CrossRef] [PubMed]
- Krug, K.; Jaehnig, E.J.; Satpathy, S.; Blumenberg, L.; Karpova, A.; Anurag, M.; Miles, G.; Mertins, P.; Geffen, Y.; Tang, L.C.; et al. Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 2020, 183, 1436–1456.e31. [Google Scholar] [CrossRef]
- Mannino, M.P.; Hart, G.W. The Beginner’s Guide to O-GlcNAc: From Nutrient Sensitive Pathway Regulation to Its Impact on the Immune System. Front. Immunol. 2022, 13, 828648. [Google Scholar] [CrossRef]
- Kissel, T.; Reijm, S.; Slot, L.M.; Cavallari, M.; Wortel, C.M.; Vergroesen, R.D.; Stoeken-Rijsbergen, G.; Kwekkeboom, J.C.; Kampstra, A.; Levarht, E.; et al. Antibodies and B Cells Recognising Citrullinated Proteins Display a Broad Cross-Reactivity towards Other Post-Translational Modifications. Ann. Rheum. Dis. 2020, 79, 472–480. [Google Scholar] [CrossRef]
- Önder, Ö.; Sidoli, S.; Carroll, M.; Garcia, B.A. Progress in Epigenetic Histone Modification Analysis by Mass Spectrometry for Clinical Investigations. Expert Rev. Proteom. 2015, 12, 499–517. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and Recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jensen, O.N. Modification-Specific Proteomics: Strategies for Characterization of Post-Translational Modifications Using Enrichment Techniques. Proteomics 2009, 9, 4632–4641. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.I.; Voshol, H.; van Oostrum, J.; Hastings, T.G.; Cascio, M.; Glucksman, M.J. Neuroproteomics: Expression Profiling of the Brain’s Proteomes in Health and Disease. Neurochem. Res. 2004, 29, 1317–1331. [Google Scholar] [CrossRef]
- Shulman, J.M.; De Jager, P.L.; Feany, M.B. Parkinson’s Disease: Genetics and Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 193–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreasen, N.; Hesse, C.; Davidsson, P.; Minthon, L.; Wallin, A.; Winblad, B.; Vanderstichele, H.; Vanmechelen, E.; Blennow, K. Cerebrospinal Fluid Beta-Amyloid(1-42) in Alzheimer Disease: Differences between Early- and Late-Onset Alzheimer Disease and Stability during the Course of Disease. Arch. Neurol. 1999, 56, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Wallin, A.; Agren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau Protein in Cerebrospinal Fluid: A Biochemical Marker for Axonal Degeneration in Alzheimer Disease? Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Vanmechelen, E.; Vanderstichele, H.; Davidsson, P.; Van Kerschaver, E.; Van Der Perre, B.; Sjögren, M.; Andreasen, N.; Blennow, K. Quantification of Tau Phosphorylated at Threonine 181 in Human Cerebrospinal Fluid: A Sandwich ELISA with a Synthetic Phosphopeptide for Standardization. Neurosci. Lett. 2000, 285, 49–52. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Hu, W.; Xie, S.; Gong, C.-X.; Iqbal, K.; Liu, F. Rapid Alteration of Protein Phosphorylation during Postmortem: Implication in the Study of Protein Phosphorylation. Sci. Rep. 2015, 5, 15709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthélemy, N.R.; Mallipeddi, N.; Moiseyev, P.; Sato, C.; Bateman, R.J. Tau Phosphorylation Rates Measured by Mass Spectrometry Differ in the Intracellular Brain vs. Extracellular Cerebrospinal Fluid Compartments and Are Differentially Affected by Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 121. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J. Expanding the Proteome: Disordered and Alternatively Folded Proteins. Q Rev. Biophys. 2011, 44, 467–518. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Sahu, M.; Srivastava, D.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Post-Translational Modifications: Regulators of Neurodegenerative Proteinopathies. Ageing Res. Rev. 2021, 68, 101336. [Google Scholar] [CrossRef]
- Wesseling, H.; Mair, W.; Kumar, M.; Schlaffner, C.N.; Tang, S.; Beerepoot, P.; Fatou, B.; Guise, A.J.; Cheng, L.; Takeda, S.; et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 2020, 183, 1699–1713.e13. [Google Scholar] [CrossRef]
- Mair, W.; Muntel, J.; Tepper, K.; Tang, S.; Biernat, J.; Seeley, W.W.; Kosik, K.S.; Mandelkow, E.; Steen, H.; Steen, J.A. FLEXITau: Quantifying Post-Translational Modifications of Tau Protein in Vitro and in Human Disease. Anal. Chem. 2016, 88, 3704–3714. [Google Scholar] [CrossRef] [Green Version]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the Ubiquitin-Proteasome System by Protein Aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The Roles of Intracellular Protein-Degradation Pathways in Neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Shattuck Lecture–Neurodegenerative Diseases and Prions. N. Engl. J. Med. 2001, 344, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Wong, E.S.P.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.-P.; Ho, M.W.L.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-Linked Ubiquitination Promotes the Formation and Autophagic Clearance of Protein Inclusions Associated with Neurodegenerative Diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A Proteomics Approach to Understanding Protein Ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Mertins, P.; Svinkina, T.; Carr, S.A. Large-Scale Identification of Ubiquitination Sites by Mass Spectrometry. Nat. Protoc. 2013, 8, 1950–1960. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global Analysis of Lysine Ubiquitination by Ubiquitin Remnant Immunoaffinity Profiling. Nat. Biotechnol. 2010, 28, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [Green Version]
- Gaunitz, S.; Tjernberg, L.O.; Schedin-Weiss, S. What Can N-Glycomics and N-Glycoproteomics of Cerebrospinal Fluid Tell Us about Alzheimer Disease? Biomolecules 2021, 11, 858. [Google Scholar] [CrossRef]
- Goyallon, A.; Cholet, S.; Chapelle, M.; Junot, C.; Fenaille, F. Evaluation of a Combined Glycomics and Glycoproteomics Approach for Studying the Major Glycoproteins Present in Biofluids: Application to Cerebrospinal Fluid. Rapid Commun. Mass Spectrom. 2015, 29, 461–473. [Google Scholar] [CrossRef]
- Gaunitz, S.; Tjernberg, L.O.; Schedin-Weiss, S. The N-Glycan Profile in Cortex and Hippocampus Is Altered in Alzheimer Disease. J. Neurochem. 2021, 159, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.G.; Veillon, L.; Mechref, Y. N-Glycan Profile of Cerebrospinal Fluids from Alzheimer’s Disease Patients Using LC-MS. J. Proteome Res. 2019, 18, 3770–3779. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Yu, Q.; Yu, Q.; Johnson, J.; Shipman, R.; Zhong, X.; Huang, J.; Asthana, S.; Carlsson, C.; Okonkwo, O.; et al. In-Depth Site-Specific Analysis of N-Glycoproteome in Human Cerebrospinal Fluid and Glycosylation Landscape Changes in Alzheimer’s Disease. Mol. Cell. Proteom. 2021, 20, 100081. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood Plasma Phosphorylated-Tau Isoforms Track CNS Change in Alzheimer’s Disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef]
- Arakhamia, T.; Lee, C.E.; Carlomagno, Y.; Duong, D.M.; Kundinger, S.R.; Wang, K.; Williams, D.; DeTure, M.; Dickson, D.W.; Cook, C.N.; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644.e12. [Google Scholar] [CrossRef]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-Glycans of Pathological Tau: Possible Occurrence of Aberrant Processing of Tau in Alzheimer’s Disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Barthélemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Perrin, R.J.; Goate, A.M.; et al. A Soluble Phosphorylated Tau Signature Links Tau, Amyloid and the Evolution of Stages of Dominantly Inherited Alzheimer’s Disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef]
- Bayoumy, S.; Verberk, I.M.W.; den Dulk, B.; Hussainali, Z.; Zwan, M.; van der Flier, W.M.; Ashton, N.J.; Zetterberg, H.; Blennow, K.; Vanbrabant, J.; et al. Clinical and Analytical Comparison of Six Simoa Assays for Plasma P-Tau Isoforms P-Tau181, P-Tau217, and P-Tau231. Alzheimer’s Res. Ther. 2021, 13, 198. [Google Scholar] [CrossRef]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits Alpha-Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of Alpha-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, P.; Öhrfelt, A.; Lashley, T.; Blennow, K.; Brinkmalm, A.; Zetterberg, H. Mass Spectrometric Analysis of Lewy Body-Enriched α-Synuclein in Parkinson’s Disease. J. Proteome Res. 2019, 18, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Foulds, P.G.; Mitchell, J.D.; Parker, A.; Turner, R.; Green, G.; Diggle, P.; Hasegawa, M.; Taylor, M.; Mann, D.; Allsop, D. Phosphorylated α-Synuclein Can Be Detected in Blood Plasma and Is Potentially a Useful Biomarker for Parkinson’s Disease. FASEB J. 2011, 25, 4127–4137. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Stewart, T.; Shi, M.; Pottiez, G.; Dator, R.; Wu, R.; Aro, P.; Schuster, R.J.; Ginghina, C.; Pan, C.; et al. An Alpha-Synuclein MRM Assay with Diagnostic Potential for Parkinson’s Disease and Monitoring Disease Progression. Proteom. Clin. Appl. 2017, 11, 1700045. [Google Scholar] [CrossRef]
- Kang, S.H.; Kim, M.E.; Jang, H.; Kwon, H.; Lee, H.; Kim, H.J.; Seo, S.W.; Na, D.L. Amyloid Positivity in the Alzheimer/Subcortical-Vascular Spectrum. Neurology 2021, 96, e2201–e2211. [Google Scholar] [CrossRef] [PubMed]
- Nutu, M.; Bourgeois, P.; Zetterberg, H.; Portelius, E.; Andreasson, U.; Parent, S.; Lipari, F.; Hall, S.; Constantinescu, R.; Hansson, O.; et al. Aβ1-15/16 as a Potential Diagnostic Marker in Neurodegenerative Diseases. Neuromolecular Med. 2013, 15, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Halim, A.; Brinkmalm, G.; Rüetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-Specific Characterization of Threonine, Serine, and Tyrosine Glycosylations of Amyloid Precursor Protein/Amyloid β-Peptides in Human Cerebrospinal Fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef] [Green Version]
- Feneberg, E.; Charles, P.D.; Finelli, M.J.; Scott, C.; Kessler, B.M.; Fischer, R.; Ansorge, O.; Gray, E.; Talbot, K.; Turner, M.R. Detection and Quantification of Novel C-terminal TDP-43 Fragments in ALS-TDP. Brain Pathol. 2021, 31, e12923. [Google Scholar] [CrossRef]
- Johnson, G.V.W.; Stoothoff, W.H. Tau Phosphorylation in Neuronal Cell Function and Dysfunction. J. Cell Sci. 2004, 117, 5721–5729. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer Disease-Specific Conformation of Hyperphosphorylated Paired Helical Filament-Tau Is Polyubiquitinated through Lys-48, Lys-11, and Lys-6 Ubiquitin Conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K. Cerebrospinal Fluid Protein Biomarkers for Alzheimer’s Disease. NeuroRx 2004, 1, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal Fluid and Plasma Biomarkers in Alzheimer Disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso, A.d.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau Pathology in Alzheimer Disease and Other Tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef]
- Alquezar, C.; Arya, S.; Kao, A.W. Tau Post-Translational Modifications: Dynamic Transformers of Tau Function, Degradation, and Aggregation. Front. Neurol. 2020, 11, 595532. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Titani, K.; Ihara, Y. Proline-Directed and Non-Proline-Directed Phosphorylation of PHF-Tau. J. Biol. Chem. 1995, 270, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Protein Sequence and Mass Spectrometric Analyses of Tau in the Alzheimer’s Disease Brain. J. Biol. Chem. 1992, 267, 17047–17054. [Google Scholar] [CrossRef]
- Hanger, D.P.; Betts, J.C.; Loviny, T.L.F.; Blackstock, W.P.; Anderton, B.H. New Phosphorylation Sites Identified in Hyperphosphorylated Tau (Paired Helical Filament-Tau) from Alzheimer’s Disease Brain Using Nanoelectrospray Mass Spectrometry. J. Neurochem. 1998, 71, 2465–2476. [Google Scholar] [CrossRef]
- Lantero-Rodriguez, J.; Snellman, A.; Benedet, A.L.; Milà-Alomà, M.; Camporesi, E.; Montoliu-Gaya, L.; Ashton, N.J.; Vrillon, A.; Karikari, T.K.; Gispert, J.D.; et al. P-Tau235: A Novel Biomarker for Staging Preclinical Alzheimer’s Disease. EMBO Mol. Med. 2021, 13, e15098. [Google Scholar] [CrossRef]
- Min, S.-W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A.; et al. Critical Role of Acetylation in Tau-Mediated Neurodegeneration and Cognitive Deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huseby, C.J.; Hoffman, C.N.; Cooper, G.L.; Cocuron, J.-C.; Alonso, A.P.; Thomas, S.N.; Yang, A.J.; Kuret, J. Quantification of Tau Protein Lysine Methylation in Aging and Alzheimer’s Disease. J. Alzheimers Dis. 2019, 71, 979–991. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Thomas, S.N.; Schafer, K.N.; Cooper, G.L.; Liao, Z.; Clark, D.J.; Yang, A.J.; Kuret, J. Lysine Methylation Is an Endogenous Post-Translational Modification of Tau Protein in Human Brain and a Modulator of Aggregation Propensity. Biochem. J. 2014, 462, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, B.D.; Love, D.C.; Hanover, J.A. O-GlcNAc Cycling: Implications for Neurodegenerative Disorders. Int. J. Biochem. Cell Biol. 2009, 41, 2134–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A Potent Mechanism-Inspired O-GlcNAcase Inhibitor That Blocks Phosphorylation of Tau in Vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Shan, X.; Macauley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc Slows Neurodegeneration and Stabilizes Tau against Aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Barthélemy, N.R.; Fenaille, F.; Hirtz, C.; Sergeant, N.; Schraen-Maschke, S.; Vialaret, J.; Buée, L.; Gabelle, A.; Junot, C.; Lehmann, S.; et al. Tau Protein Quantification in Human Cerebrospinal Fluid by Targeted Mass Spectrometry at High Sequence Coverage Provides Insights into Its Primary Structure Heterogene.eity. J. Proteome Res. 2016, 15, 667–676. [Google Scholar] [CrossRef]
- Sato, C.; Barthélemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e7. [Google Scholar] [CrossRef] [Green Version]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal Fluid P-Tau217 Performs Better than p-Tau181 as a Biomarker of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef] [Green Version]
- Barthélemy, N.R.; Bateman, R.J.; Marin, P.; Becher, F.; Sato, C.; Lehmann, S.; Gabelle, A. Tau Hyperphosphorylation on T217 in Cerebrospinal Fluid Is Specifically Associated to Amyloid-β Pathology. bioRxiv 2017, 226977. [Google Scholar] [CrossRef] [Green Version]
- Viodé, A.; Epelbaum, S.; Benyounes, I.; Verny, M.; Dubois, B.; Junot, C.; Fenaille, F.; Lamari, F.; Becher, F. Simultaneous Quantification of Tau and α-Synuclein in Cerebrospinal Fluid by High-Resolution Mass Spectrometry for Differentiation of Lewy Body Dementia from Alzheimer’s Disease and Controls. Analyst 2019, 144, 6342–6351. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Limanaqi, F.; Busceti, C.L.; Mastroiacovo, F.; Nicoletti, F.; Puglisi-Allegra, S.; Fornai, F. Glymphatic System as a Gateway to Connect Neurodegeneration From Periphery to CNS. Front. Neurosci. 2021, 15, 639140. [Google Scholar] [CrossRef] [PubMed]
- Cicognola, C.; Brinkmalm, G.; Wahlgren, J.; Portelius, E.; Gobom, J.; Cullen, N.C.; Hansson, O.; Parnetti, L.; Constantinescu, R.; Wildsmith, K.; et al. Novel Tau Fragments in Cerebrospinal Fluid: Relation to Tangle Pathology and Cognitive Decline in Alzheimer’s Disease. Acta Neuropathol. 2019, 137, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.C. The Role of α-Synuclein in Neurodegenerative Diseases. Pharmacol. Ther. 2005, 105, 311–331. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Lee, V.M.-Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s Disease Linked to Pathological Alpha-Synuclein: New Targets for Drug Discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. Alpha-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of Alpha-Synuclein in Lewy Bodies of Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative Measurement of Intact Alpha-Synuclein Proteoforms from Post-Mortem Control and Parkinson’s Disease Brain Tissue by Intact Protein Mass Spectrometry. Sci. Rep. 2014, 4, 5797. [Google Scholar] [CrossRef]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full Length Alpha-Synuclein Is Present in Cerebrospinal Fluid from Parkinson’s Disease and Normal Subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.; et al. Alpha-Synuclein Implicated in Parkinson’s Disease Is Present in Extracellular Biological Fluids, Including Human Plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Metzger, F.; Nagl, M.; von Arnim, C.A.F.; Halbgebauer, S.; Steinacker, P.; Ludolph, A.C.; Otto, M. Alpha-, Beta-, and Gamma-Synuclein Quantification in Cerebrospinal Fluid by Multiple Reaction Monitoring Reveals Increased Concentrations in Alzheimer’s and Creutzfeldt-Jakob Disease but No Alteration in Synucleinopathies. Mol. Cell. Proteom. 2016, 15, 3126–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; von Arnim, C.A.F.; Diehl-Schmid, J.; Froelich, L.; Grimmer, T.; Hausner, L.; Denk, J.; Jahn, H.; et al. Targeted Mass Spectrometry Suggests Beta-Synuclein as Synaptic Blood Marker in Alzheimer’s Disease. J. Proteome Res. 2020, 19, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Pero-Gascon, R.; Benavente, F.; Minic, Z.; Berezovski, M.V.; Sanz-Nebot, V. On-Line Aptamer Affinity Solid-Phase Extraction Capillary Electrophoresis-Mass Spectrometry for the Analysis of Blood α-Synuclein. Anal. Chem. 2020, 92, 1525–1533. [Google Scholar] [CrossRef]
- Wildburger, N.C.; Esparza, T.J.; LeDuc, R.D.; Fellers, R.T.; Thomas, P.M.; Cairns, N.J.; Kelleher, N.L.; Bateman, R.J.; Brody, D.L. Diversity of Amyloid-Beta Proteoforms in the Alzheimer’s Disease Brain. Sci. Rep. 2017, 7, 9520. [Google Scholar] [CrossRef] [Green Version]
- Brinkmalm, G.; Portelius, E.; Öhrfelt, A.; Mattsson, N.; Persson, R.; Gustavsson, M.K.; Vite, C.H.; Gobom, J.; Månsson, J.-E.; Nilsson, J.; et al. An Online Nano-LC-ESI-FTICR-MS Method for Comprehensive Characterization of Endogenous Fragments from Amyloid β and Amyloid Precursor Protein in Human and Cat Cerebrospinal Fluid. J. Mass Spectrom. 2012, 47, 591–603. [Google Scholar] [CrossRef]
- Kametani, F.; Obi, T.; Shishido, T.; Akatsu, H.; Murayama, S.; Saito, Y.; Yoshida, M.; Hasegawa, M. Mass Spectrometric Analysis of Accumulated TDP-43 in Amyotrophic Lateral Sclerosis Brains. Sci. Rep. 2016, 6, 23281. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Kasai, T.; Kojima, Y.; Ohmichi, T.; Tatebe, H.; Tsuji, Y.; Noto, Y.-I.; Kitani-Morii, F.; Shinomoto, M.; Allsop, D.; Mizuno, T.; et al. Combined Use of CSF NfL and CSF TDP-43 Improves Diagnostic Performance in ALS. Ann. Clin. Transl. Neurol. 2019, 6, 2489–2502. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Ye, M.; Zou, H. Enrichment and Separation Techniques for Large-Scale Proteomics Analysis of the Protein Post-Translational Modifications. J. Chromatogr. A. 2014, 1372, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Eng, J.; Yates, J.R.; Tollaksen, S.L.; Giometti, C.S.; Holden, J.F.; Adams, M.W.W.; Reich, C.I.; Olsen, G.J.; Hays, L.G. Identification of 2D-Gel Proteins: A Comparison of MALDI/TOF Peptide Mass Mapping to Mu LC-ESI Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom 2003, 14, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible Workflow for Multiplexed Deep-Scale Proteome and Phosphoproteome Analysis of Tumor Tissues by Liquid Chromatography-Mass Spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Prus, G.; Hoegl, A.; Weinert, B.T.; Choudhary, C. Analysis and Interpretation of Protein Post-Translational Modification Site Stoichiometry. Trends Biochem. Sci. 2019, 44, 943–960. [Google Scholar] [CrossRef] [PubMed]
- Siuti, N.; Kelleher, N.L. Decoding Protein Modifications Using Top-down Mass Spectrometry. Nat. Methods 2007, 4, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Leney, A.C.; Heck, A.J.R. Native Mass Spectrometry: What Is in the Name? J. Am. Soc. Mass Spectrom 2017, 28, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642. [Google Scholar] [CrossRef]
- Simithy, J.; Sidoli, S.; Garcia, B.A. Integrating Proteomics and Targeted Metabolomics to Understand Global Changes in Histone Modifications. Proteomics 2018, 18, e1700309. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo, R.; Jacquemin, C.; Villain, N.; Fenaille, F.; Lamari, F.; Becher, F. Mass Spectrometry for Neurobiomarker Discovery: The Relevance of Post-Translational Modifications. Cells 2022, 11, 1279. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11081279
Azevedo R, Jacquemin C, Villain N, Fenaille F, Lamari F, Becher F. Mass Spectrometry for Neurobiomarker Discovery: The Relevance of Post-Translational Modifications. Cells. 2022; 11(8):1279. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11081279
Chicago/Turabian StyleAzevedo, Rita, Chloé Jacquemin, Nicolas Villain, François Fenaille, Foudil Lamari, and François Becher. 2022. "Mass Spectrometry for Neurobiomarker Discovery: The Relevance of Post-Translational Modifications" Cells 11, no. 8: 1279. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11081279