
Circulated TGF-β1 and VEGF-A as Biomarkers for Fabry Disease-Associated Cardiomyopathy


Abstract
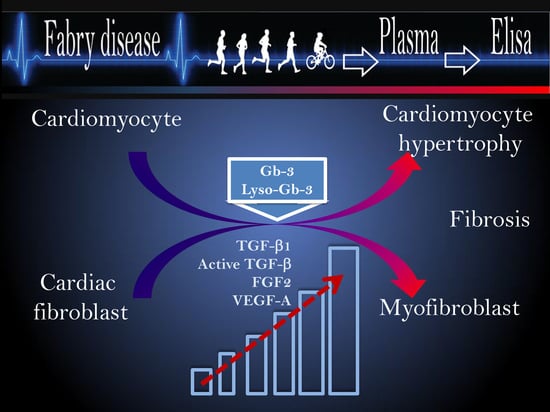
:
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Enzyme-Linked Immunosorbent Assay (ELISA):
2.3. Statistical Analysis
3. Results
3.1. Circulating Levels of TGF-β1 and the Active Form of TGF-β1 Are Elevated in Patients with Fabry Disease
3.2. Circulating Angiogenic Biomarkers FGF2 and VEGF-A Are Elevated in FD
3.3. The Association between Growth Factors and Lyso-Gb-3 Accumulation in FD
3.4. Plasma TGF-β1 Is Elevated in FD Patients with Abnormal EKG and HCM
3.5. Active TGF-β Does Not Correlate with Cardiomyopathy in Fabry Disease
3.6. The Association of VEGF-A and FGF2 with FD Cardiomyopathy
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Adusumilli, G.; Kaggie, J.D.; D’Amore, S.; Cox, T.M.; Deegan, P.; MacKay, J.W.; McDonald, S.; Consortium, G. Improving the quantitative classification of Erlenmeyer flask deformities. Skelet. Radiol. 2021, 50, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Sanchez-Nino, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Touboul, D.; Roy, S.; Germain, D.P.; Baillet, A.; Brion, F.; Prognon, P.; Chaminade, P.; Laprevote, O. Fast fingerprinting by MALDI-TOF mass spectrometry of urinary sediment glycosphingolipids in Fabry disease. Anal. Bioanal. Chem. 2005, 382, 1209–1216. [Google Scholar] [CrossRef]
- Boutin, M.; Menkovic, I.; Martineau, T.; Vaillancourt-Lavigueur, V.; Toupin, A.; Auray-Blais, C. Separation and Analysis of Lactosylceramide, Galabiosylceramide, and Globotriaosylceramide by LC-MS/MS in Urine of Fabry Disease Patients. Anal. Chem. 2017, 89, 13382–13390. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [PubMed]
- Machann, W.; Breunig, F.; Weidemann, F.; Sandstede, J.; Hahn, D.; Kostler, H.; Neubauer, S.; Wanner, C.; Beer, M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur. J. Heart Fail. 2011, 13, 278–283. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef]
- Mauhin, W.; Lidove, O.; Masat, E.; Mingozzi, F.; Mariampillai, K.; Ziza, J.M.; Benveniste, O. Innate and Adaptive Immune Response in Fabry Disease. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 22, pp. 1–10. [Google Scholar]
- Liu, G.; Ma, C.; Yang, H.; Zhang, P.Y. Transforming growth factor beta and its role in heart disease. Exp. Ther. Med. 2017, 13, 2123–2128. [Google Scholar] [CrossRef]
- Almendral, J.L.; Shick, V.; Rosendorff, C.; Atlas, S.A. Association between transforming growth factor-beta(1) and left ventricular mass and diameter in hypertensive patients. J. Am. Soc. Hypertens. 2010, 4, 135–141. [Google Scholar] [CrossRef]
- Ayca, B.; Sahin, I.; Kucuk, S.H.; Akin, F.; Kafadar, D.; Avsar, M.; Avci, I.I.; Gungor, B.; Okuyan, E.; Dinckal, M.H. Increased Transforming Growth Factor-beta Levels Associated With Cardiac Adverse Events in Hypertrophic Cardiomyopathy. Clin. Cardiol. 2015, 38, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-beta in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-beta Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Khurana, R.; Simons, M.; Martin, J.F.; Zachary, I.C. Role of angiogenesis in cardiovascular disease: A critical appraisal. Circulation 2005, 112, 1813–1824. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Mauhin, W.; Abily-Donval, L.; Lesueur, C.; Berger, M.G.; Nadjar, Y.; Berger, J.; Benveniste, O.; Lamari, F.; Laforet, P.; et al. A Proteomics-Based Analysis Reveals Predictive Biological Patterns in Fabry Disease. J. Clin. Med. 2020, 9, 1325. [Google Scholar] [CrossRef]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-beta(1) (Transforming Growth Factor-beta(1)) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythmia Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef]
- Levstek, T.; Vujkovac, B.; Trebusak Podkrajsek, K. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes 2020, 11, 1091. [Google Scholar] [CrossRef]
- Nowak, A.; Mechtler, T.P.; Desnick, R.J.; Kasper, D.C. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol. Genet. Metab. 2017, 120, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Sakuraba, H.; Togawa, T.; Tsukimura, T.; Kato, H. Plasma lyso-Gb3: A biomarker for monitoring fabry patients during enzyme replacement therapy. Clin. Exp. Nephrol. 2018, 22, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Hagege, A.; Reant, P.; Habib, G.; Damy, T.; Barone-Rochette, G.; Soulat, G.; Donal, E.; Germain, D.P. Fabry disease in cardiology practice: Literature review and expert point of view. Arch. Cardiovasc. Dis. 2019, 112, 278–287. [Google Scholar] [CrossRef]
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC Cardiovasc. Imaging 2019, 12 Pt 2, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Vardarli, I.; Weber, M.; Rischpler, C.; Fuhrer, D.; Herrmann, K.; Weidemann, F. Fabry Cardiomyopathy: Current Treatment and Future Options. J. Clin. Med. 2021, 10, 3026. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, O.; Cordeiro, F.; Gago, M.F.; Miltenberger-Miltenyi, G.; Ferreira, C.; Sousa, N.; Cunha, D. Fabry Disease and the Heart: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4434. [Google Scholar] [CrossRef] [PubMed]
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ. Cardiovasc. Imaging 2013, 6, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Umer, M.; Motwani, M.; Jefferies, J.L.; Nagueh, S.F.; Kalra, D.K. Cardiac Involvement in Fabry Disease and the Role of Multimodality Imaging in Diagnosis and Disease Monitoring. Curr. Probl. Cardiol. 2023, 48, 101439. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Villar, A.V.; Cobo, M.; Llano, M.; Montalvo, C.; Gonzalez-Vilchez, F.; Martin-Duran, R.; Hurle, M.A.; Nistal, J.F. Plasma levels of transforming growth factor-beta1 reflect left ventricular remodeling in aortic stenosis. PLoS ONE 2009, 4, e8476. [Google Scholar] [CrossRef] [PubMed]
- Glazer, N.L.; Macy, E.M.; Lumley, T.; Smith, N.L.; Reiner, A.P.; Psaty, B.M.; King, G.L.; Tracy, R.P.; Siscovick, D.S. Transforming growth factor beta-1 and incidence of heart failure in older adults: The Cardiovascular Health Study. Cytokine 2012, 60, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Zampetti, A.; Gnarra, M.; Borsini, W.; Giurdanella, F.; Antuzzi, D.; Piras, A.; Smaldone, C.; Pieroni, M.; Cadeddu, C.; de Waure, C.; et al. Vascular endothelial growth factor (VEGF-a) in Fabry disease: Association with cutaneous and systemic manifestations with vascular involvement. Cytokine 2013, 61, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Tufro, A.; Veron, D. VEGF and podocytes in diabetic nephropathy. Semin. Nephrol. 2012, 32, 385–393. [Google Scholar] [CrossRef]
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461. [Google Scholar] [CrossRef]
- Weidemann, F.; Breunig, F.; Beer, M.; Sandstede, J.; Stork, S.; Voelker, W.; Ertl, G.; Knoll, A.; Wanner, C.; Strotmann, J.M. The variation of morphological and functional cardiac manifestation in Fabry disease: Potential implications for the time course of the disease. Eur. Heart J. 2005, 26, 1221–1227. [Google Scholar] [CrossRef]
- Sheppard, M.N.; Cane, P.; Florio, R.; Kavantzas, N.; Close, L.; Shah, J.; Lee, P.; Elliott, P. A detailed pathologic examination of heart tissue from three older patients with Anderson-Fabry disease on enzyme replacement therapy. Cardiovasc. Pathol. 2010, 19, 293–301. [Google Scholar] [CrossRef]
- Nowak, A.; Beuschlein, F.; Sivasubramaniam, V.; Kasper, D.; Warnock, D.G. Lyso-Gb3 associates with adverse long-term outcome in patients with Fabry disease. J. Med. Genet. 2022, 59, 287–293. [Google Scholar] [CrossRef]
- Maruyama, H.; Miyata, K.; Mikame, M.; Taguchi, A.; Guili, C.; Shimura, M.; Murayama, K.; Inoue, T.; Yamamoto, S.; Sugimura, K.; et al. Effectiveness of plasma lyso-Gb3 as a biomarker for selecting high-risk patients with Fabry disease from multispecialty clinics for genetic analysis. Genet. Med. 2019, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Nino, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy. Nephrol. Dial. Transplant. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Jung, N.; Park, J.W.; Park, H.Y.; Jung, S.C. Epithelial-Mesenchymal Transition in Kidney Tubular Epithelial Cells Induced by Globotriaosylsphingosine and Globotriaosylceramide. PLoS ONE 2015, 10, e0136442. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Choi, E.N.; Jeon, Y.J.; Jung, S.C. Possible role of transforming growth factor-beta1 and vascular endothelial growth factor in Fabry disease nephropathy. Int. J. Mol. Med. 2012, 30, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF-A: A critical regulator of blood vessel growth. Eur. Cytokine Netw. 2009, 20, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.A.; Zwetsloot, K.A.; Westerkamp, L.M.; Hickner, R.C.; Pofahl, W.E.; Gavin, T.P. Lower skeletal muscle capillarization and VEGF expression in aged vs. young men. J. Appl. Physiol. 2006, 100, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Yusifov, A.; Woulfe, K.C.; Bruns, D.R. Mechanisms and implications of sex differences in cardiac aging. J. Cardiovasc. Aging 2022, 2, 20. [Google Scholar] [CrossRef]
- Kessler, E.L.; Rivaud, M.R.; Vos, M.A.; van Veen, T.A.B. Sex-specific influence on cardiac structural remodeling and therapy in cardiovascular disease. Biol. Sex Differ. 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, K.; Ichimura, K.; Reddy, S.; Haddad, F.; Spiekerkoetter, E. Cardiac Fibrosis in the Pressure Overloaded Left and Right Ventricle as a Therapeutic Target. Front. Cardiovasc. Med. 2022, 9, 886553. [Google Scholar] [CrossRef]
- Lin, C.Y.; Chung, F.P.; Lin, Y.J.; Chang, S.L.; Lo, L.W.; Hu, Y.F.; Tuan, T.C.; Chao, T.F.; Liao, J.N.; Chang, Y.T.; et al. Gender differences in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: Clinical manifestations, electrophysiological properties, substrate characteristics, and prognosis of radiofrequency catheter ablation. Int. J. Cardiol. 2017, 227, 930–937. [Google Scholar] [CrossRef]
- Niemann, M.; Herrmann, S.; Hu, K.; Breunig, F.; Strotmann, J.; Beer, M.; Machann, W.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Differences in Fabry cardiomyopathy between female and male patients: Consequences for diagnostic assessment. JACC Cardiovasc. Imaging 2011, 4, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Aerts, J.M.; Auray-Blais, C.; Maruyama, H.; Mehta, A.B.; Skuban, N.; Krusinska, E.; Schiffmann, R. Assessment of plasma lyso-Gb(3) for clinical monitoring of treatment response in migalastat-treated patients with Fabry disease. Genet. Med. 2021, 23, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Talbot, A.; Nicholls, K.; Fletcher, J.M.; Fuller, M. A simple method for quantification of plasma globotriaosylsphingosine: Utility for Fabry disease. Mol. Genet. Metab. 2017, 122, 121–125. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Age (Years) | Sex M/F | Genotype (Allele 1/Allee2) | Plasma Lyso-Gb-3 ng/mL | Urine Lyso-Gb3 µg/mmol/cr | Therapy (Years) |
|---|---|---|---|---|---|---|
| FD subjects without cardiomyopathy | ||||||
| 1 | 25 | F | c.718_719del | 0.46 | 8.3 | * ERT > 10 |
| 2 | 45 | F | C223Y | 2.9 | 70 | ERT 5–10 |
| 3 | 46 | F | c.966DelC | - | - | ERT < 5 |
| 4 | 21 | F | R49P | 2.9 | <6 | ERT > 10 |
| 5 | 18 | F | R363 H | 0.3 | 8.6 | ERT < 5; CT-2; ERT 2 |
| 6 | 27 | F | c1033_1034delTC | 1.4 | 220 | * ERT > 10 |
| 7 | 33 | F | c.1072_1074del | 6.9 | 151 | naïve |
| 8 | 28 | M | 422C>T | 11 | 41 | ERT > 10 |
| 9 | 18 | M | R49P | 11 | 19 | * ERT > 10 |
| 10 | 36 | M | M296V | 0.84 | 102 | ERT 5 |
| 11 | 53 | M | A143T | 0.39 | 14 | CT > 10 |
| 12 | 39 | F | N215S | 0.3 | 18 | ERT-2; CT-4 |
| 13 | 56 | F | G328R | 5.2 | 15 | * ERT > 10 |
| 14 | 68 | F | C2233Y | 4.5 | 31 | ERT < 5 |
| 15 | 26 | M | c.718_719delAA | 13 | 825 | ERT < 5 |
| 16 | 58 | M | T41I | 0.4 | 13 | CT < 5 |
| 17 | 42 | M | N215S | 1.1 | 57 | ERT > 10 |
| FD subjects with hypertrophic cardiomyopathy (HCM) | ||||||
| 18 | 57 | F | R49P | 4.9 | 40 | * ERT > 10 |
| 19 | 41 | F | R301X | 11 | 63 | ERT < 5 |
| 20 | 46 | F | c.718_719delAA | 2.9 | - | *ERT > 10 |
| 21 | 46 | F | Q279E | 2.1 | 32 | CT < 5 |
| 22 | 44 | F | Arg227Gln | 2.9 | 17 | ERT < 5 |
| 23 | 21 | F | c1033_1034delTC | 2.1 | 84 | CT < 5 |
| 24 | 22 | F | D244N | 5.3 | 96 | *ERT 5–10 |
| 25 | 42 | F | G325D | 2.6 | 29 | ERT > 10 |
| 26 | 62 | F | Gly325Asp | 2.7 | 17 | ERT > 10 |
| 27 | 40 | F | A143T | 0.3 | 141 | CT < 5 |
| 28 | 59 | F | R118C | 0.1 | 101 | naive |
| 29 | 24 | M | W277X | 18 | 620 | ERT 5–10 |
| 30 | 42 | M | R227Q | 15 | 92 | ERT > 10 |
| 31 | 34 | M | V296E | 14 | 1107 | *ERT > 10 |
| 32 | 26 | M | G325D | 17 | 177 | * ERT > 10 |
| 33 | 20 | M | G325D | 5.3 | 8.7 | ERT < 5 |
| 34 | 25 | M | Y134D | 6.9 | 6.4 | ERT > 10 |
| 35 | 53 | M | Y207C | 12 | 413 | * ERT > 10 |
| 36 | 65 | F | N215S | 0.6 | 24 | ERT 5–10 |
| 37 | 40 | F | c.718_719del | 0.3 | 21 | ERT >10 |
| 38 | 45 | F | R363H | 0.3 | 21 | ERT 5–10 |
| 39 | 23 | M | c.966DelC | - | - | ERT 5–10 |
| 40 | 43 | M | c1033_1034delTC | 77 | 1033 | ERT < 5 y |
| 41 | 34 | M | A143T | 0.9 | 34 | ERT < 5 |
| 42 | 65 | M | C223Y | 20 | 231 | ERT > 10 |
| 43 | 58 | M | c.777del | 17 | 33 | * ERT > 10 |
| 44 | 58 | M | c.717_718del Frameshift | 19 | 1257 | * ERT > 10 |
| 45 | 48 | M | c.1072_1074del | 12 | 38 | ERT 5–10 |
| No HCM | Borderline | HCM Mild | HCM Moderate/Severe | |||||
|---|---|---|---|---|---|---|---|---|
| F (n = 7) | M (n = 4) | F (n = 3) | M (n = 3) | F (n = 11) | M (n = 7) | F (n = 3) | M (n = 7) | |
| Age | 30.7 ± 11 | 33.4 ± 14 | 54 ± 14 | 42.6 ± 15 | 43.6 ± 13 | 32 ± 12 | 50 ± 13 | 47 ± 14 |
| Age min | 18 | 18 | 39 | 27 | 22 | 21 | 40 | 24 |
| Age max | 46 | 53 | 68 | 58 | 63 | 54 | 65 | 65 |
| LVPWd | 0.7 ± 0.1 | 0.8 ± 0.05 | 0.8 ± 0.1 | 0.9 ± 0.06 | 1.08 ± 0.08 | 1.1 ± 0.05 | 1.3 ± 0.15 | 1.4 ± 0.1 |
| LV mass | 84.6 ± 16 | 116.6 ± 17 | 83 ± 4 | 120 ± 11 | 118.1 ± 29 | 173 ± 31 | 243 | 282 ± 169 |
| LVEF % | 62.3 ± 3 | 61.5 ± 2 | 56.3 ± 2 | 62.6 ± 5 | 59.3 ± 3 | 57.1 ± 5 | 54.0 ± 8 | 54 ± 3 |
| EKG | normal | normal | abnormal | abnormal | abnormal | abnormal | abnormal | abnormal |
| Fibrosis | no | No | no | no | 5 | 2 | 1 & | 5 |
| Biomarker | CNT vs. FD | CNT vs. FD Females | CNT vs. FD Males | |||
|---|---|---|---|---|---|---|
| FD | CNT | FD | CNT | FD | CNT | |
| TGFβ1 95% CI | 38.7 ± 4.1 | 12.5 ± 1.4 | 34.7 ± 4.7 | 13.9 ± 1.8 | 43.4 ± 7.1 | 10.9 ± 2.3 |
| T test p < 0.0001 17.3 to 35 | T test p < 0.0001 10.4 to 31 | T test p < 0.0001 16.9 to 47.9 | ||||
| Active TGFβ 95% CI | 148 ± 11 | 111 ± 15 | 125 ± 16 | 81.93 ± 13 | 174 ± 16 | 145 ± 28 |
| T test p < 0.05 −2.2 to 74.8 | T test p < 0.05 −1.1 to 87.2 | T test p = 0.1 | ||||
| VEGF-A 95% CI | 217.3 ± 54.6 | 71.4 ± 9.5 | 163.8 ± 62.4 | 64.7 ± 13.2 | 278.4 ± 92 | 79.6 ± 14 |
| T test p < 0.005 26.1 to 265.7 | T test p = 0.11, F test p < 0.0001 | T test p < 0.05 −12 to 410 | ||||
| FGF2 95% CI | 90.7 ± 8.9 | 63.3 ± 6.8 | 95.2 ± 15 | 57.3 ± 6.2 | 85.8 ± 8.5 | 69.5 ± 12.9 |
| T test p < 0.005 4.8 to 50.1 | T test p < 0.05 3.1 to 72.5 | F test p = 0.14 | ||||
| Plasma Lyso-Gb-3 | Urine Lyso-Gb-3 | |||
|---|---|---|---|---|
| Significant | Pearson Correlation | Significant | Pearson Correlation | |
| TGFβ1 | Yes | * p = 0.02, R = 0.3 | Yes | * p = 0.007, R = 0.4 |
| Active TGFβ | No | Yes | * p = 0.02, R = 0.3 | |
| VEGF-A | Yes | ** p = 0.005, R = 0.3 | No | |
| FGF2 | No | No | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, M.M.; Dao, J.; Slayeh, O.A.; Friedman, A.; Goker-Alpan, O. Circulated TGF-β1 and VEGF-A as Biomarkers for Fabry Disease-Associated Cardiomyopathy. Cells 2023, 12, 2102. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12162102
Ivanova MM, Dao J, Slayeh OA, Friedman A, Goker-Alpan O. Circulated TGF-β1 and VEGF-A as Biomarkers for Fabry Disease-Associated Cardiomyopathy. Cells. 2023; 12(16):2102. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12162102
Chicago/Turabian StyleIvanova, Margarita M., Julia Dao, Omar Abu Slayeh, Andrew Friedman, and Ozlem Goker-Alpan. 2023. "Circulated TGF-β1 and VEGF-A as Biomarkers for Fabry Disease-Associated Cardiomyopathy" Cells 12, no. 16: 2102. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12162102