
When You Come to a Fork in the Road, Take It: Wnt Signaling Activates Multiple Pathways through the APC/Axin/GSK-3 Complex

Abstract
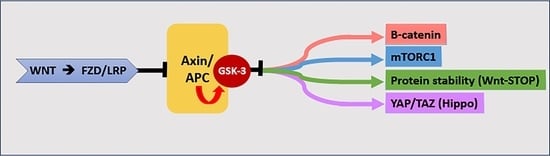
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. General Features of GSK-3
2.1. GSK-3α and GSK-3β Are Closely Related Serine/Threonine Kinases
2.2. Distinct Functions of GSK-3α and GSK-3β
2.3. A Multitude of GSK-3 Targets
2.4. GSK-3 Frequently Functions within a Scaffold Complex
2.5. GSK-3 Is Constitutively Active and Is Inhibited by Diverse Signals
2.6. GSK-3 Has Multiple Functions in Addition to Its Roles in Wnt and RTK Signaling
3. Wnt Signaling
3.1. Wnt/β Catenin Pathway
3.2. When You Come to a Fork in the Road, Take It [100]: Divergence in Canonical Wnt Signaling
3.3. Wnt/GSK-3 Signaling in Cancer
4. Summary
Funding
Acknowledgments
Conflicts of Interest
References
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011, 505607. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Hoeflich, K.P.; Woodgett, J.R. Glycogen synthase kinase-3: Properties, functions, and regulation. Chem. Rev. 2001, 101, 2527–2540. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Klein, P.S. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front. Mol. Neurosci. 2011, 5, 1. [Google Scholar] [CrossRef]
- Snitow, M.E.; Bhansali, R.S.; Klein, P.S. Lithium and Therapeutic Targeting of GSK-3. Cells 2021, 10, 255. [Google Scholar] [CrossRef]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; Maestro, R.; et al. Multifaceted roles of GSK-3 and Wnt/beta-catenin in hematopoiesis and leukemogenesis: Opportunities for therapeutic intervention. Leukemia 2014, 28, 15–33. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Yellowlees, D.; Kernohan, J.C.; Cohen, P. Purification of glycogen synthase kinase 3 from rabbit skeletal muscle. Copurification with the activating factor (FA) of the (Mg-ATP) dependent protein phosphatase. Eur. J. Biochem. FEBS 1981, 119, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. How to continually make the case for fundamental science: From the perspective of a protein kinase. Biochem. Cell Biol. 2019, 97, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.; Hayes, J.D.; Sutherland, C. A partnership with the proteasome; the destructive nature of GSK3. Biochem. Pharmacol. 2018, 147, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Ruel, L.; Woodgett, J. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1668. [Google Scholar] [CrossRef]
- Domoto, T.; Uehara, M.; Bolidong, D.; Minamoto, T. Glycogen Synthase Kinase 3beta in Cancer Biology and Treatment. Cells 2020, 9, 1388. [Google Scholar] [CrossRef]
- Duda, P.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Martelli, A.M.; Cocco, L.; Ratti, S.; Candido, S.; Libra, M.; Montalto, G.; et al. Targeting GSK3 and Associated Signaling Pathways Involved in Cancer. Cells 2020, 9, 1110. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Paganelli, F.; Chiarini, F.; McCubrey, J.A. GSK-3: A multifaceted player in acute leukemias. Leukemia 2021, 35, 1829–1842. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Rim, E.Y.; Clevers, H.; Nusse, R. The Wnt Pathway: From Signaling Mechanisms to Synthetic Modulators. Annu. Rev. Biochem. 2022, 91, 571–598. [Google Scholar] [CrossRef]
- Nusse, R. The Wnt Gene Homepage. 1999. Available online: http://www.stanford.edu/~rnusse/wntwindow.html (accessed on 15 August 2023).
- Koca, Y.; Collu, G.M.; Mlodzik, M. Wnt-frizzled planar cell polarity signaling in the regulation of cell motility. Curr. Top. Dev. Biol. 2022, 150, 255–297. [Google Scholar] [CrossRef]
- Shi, D.L. Wnt/planar cell polarity signaling controls morphogenetic movements of gastrulation and neural tube closure. Cell Mol. Life Sci. 2022, 79, 586. [Google Scholar] [CrossRef]
- Wallingford, J.B.; Mitchell, B. Strange as it may seem: The many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011, 25, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.T.; Wallingford, J.B. Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef]
- Cole, A.; Frame, S.; Cohen, P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 2004, 377, 249–255. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. Embo J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Alon, L.T.; Pietrokovski, S.; Barkan, S.; Avrahami, L.; Kaidanovich-Beilin, O.; Woodgett, J.R.; Barnea, A.; Eldar-Finkelman, H. Selective loss of glycogen synthase kinase-3alpha in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 2011, 585, 1158–1162. [Google Scholar] [CrossRef]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Lipina, T.V.; Takao, K.; van Eede, M.; Hattori, S.; Laliberté, C.; Khan, M.; Okamoto, K.; Chambers, J.W.; Fletcher, P.J.; et al. Abnormalities in brain structure and behavior in GSK-3alpha mutant mice. Mol. Brain 2009, 2, 35. [Google Scholar] [CrossRef]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF- kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.J.; Arron, J.R.; Stankunas, K.; Crabtree, G.R.; Longaker, M.T. Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature 2007, 446, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional redundancy of GSK-3alpha and GSK-3beta in Wnt/beta-catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Goswami, S.; Dey, S.; Gangoda, M.; Brothag, C.; Eisa, A.; Woodgett, J.; Phiel, C.; Kline, D.; Vijayaraghavan, S. Isoform-specific requirement for GSK3alpha in sperm for male fertility. Biol. Reprod. 2018, 99, 384–394. [Google Scholar] [CrossRef]
- Zhou, J.; Freeman, T.A.; Ahmad, F.; Shang, X.; Mangano, E.; Gao, E.; Farber, J.; Wang, Y.; Ma, X.L.; Woodgett, J.; et al. GSK-3alpha is a central regulator of age-related pathologies in mice. J. Clin. Investig. 2013, 123, 1821–1832. [Google Scholar] [CrossRef]
- Park, D.S.; Nguyen, S.C.; Isenhart, R.; Shah, P.P.; Kim, W.; Barnett, R.J.; Chandra, A.; Luppino, J.M.; Harke, J.; Wai, M.; et al. High-throughput Oligopaint screen identifies druggable 3D genome regulators. Nature 2023, 620, 209–217. [Google Scholar] [CrossRef]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3alpha as a target in human acute myeloid leukemia. J. Clin. Investig. 2012, 122, 935–947. [Google Scholar] [CrossRef]
- Neumann, T.; Benajiba, L.; Goring, S.; Stegmaier, K.; Schmidt, B. Evaluation of Improved Glycogen Synthase Kinase-3alpha Inhibitors in Models of Acute Myeloid Leukemia. J. Med. Chem. 2015, 58, 8907–8919. [Google Scholar] [CrossRef]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective inhibition of glycogen synthase kinase 3alpha corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572. [Google Scholar] [CrossRef] [PubMed]
- Soutar, M.P.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 2010, 115, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Lu, Y.; Chen, H.N.; Li, Z.; Hu, M.; Zhang, R.; Wang, X.; Xu, Z.; Gong, Y.; Wang, R.; et al. Deciphering the Clinical Significance and Kinase Functions of GSK3alpha in Colon Cancer by Proteomics and Phosphoproteomics. Mol. Cell Proteom. 2023, 22, 100545. [Google Scholar] [CrossRef]
- Liu, X.; Salokas, K.; Tamene, F.; Jiu, Y.; Weldatsadik, R.G.; Ohman, T.; Varjosalo, M. An AP-MS- and BioID-compatible MAC-tag enables comprehensive mapping of protein interactions and subcellular localizations. Nat. Commun. 2018, 9, 1188. [Google Scholar] [CrossRef] [PubMed]
- Cormier, K.W.; Larsen, B.; Gingras, A.C.; Woodgett, J.R. Interactomes of Glycogen Synthase Kinase-3 Isoforms. J. Proteome Res. 2023, 22, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Yellowlees, D.; Aitken, A.; Donella-Deana, A.; Hemmings, B.A.; Parker, P.J. Separation and characterisation of glycogen synthase kinase 3, glycogen synthase kinase 4 and glycogen synthase kinase 5 from rabbit skeletal muscle. Eur. J. Biochem. FEBS 1982, 124, 21–35. [Google Scholar] [CrossRef]
- DePaoli-Roach, A.A.; Roach, P.J.; Larner, J. Multiple phosphorylation of rabbit skeletal muscle glycogen synthase. Comparison of the actions of different protein kinases capable of catalyzing phosphorylation in vitro. J. Biol. Chem. 1979, 254, 12062–12068. [Google Scholar] [CrossRef] [PubMed]
- Fiol, C.J.; Wang, A.; Roeske, R.W.; Roach, P.J. Ordered multisite protein phosphorylation. Analysis of glycogen synthase kinase 3 action using model peptide substrates. J. Biol. Chem. 1990, 265, 6061–6065. [Google Scholar] [CrossRef]
- Fiol, C.J.; Mahrenholz, A.M.; Wang, Y.; Roeske, R.W.; Roach, P.J. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem. 1987, 262, 14042–14048. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P.; Biondi, R.M. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shinde, M.Y.; Sidoli, S.; Kulej, K.; Mallory, M.J.; Radens, C.M.; Reicherter, A.L.; Myers, R.L.; Barash, Y.; Lynch, K.W.; Garcia, B.A.; et al. Phosphoproteomics reveals that glycogen synthase kinase-3 phosphorylates multiple splicing factors and is associated with alternative splicing. J. Biol. Chem. 2017, 292, 18240–18255. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Heyd, F.; Lynch, K.W. Phosphorylation-dependent regulation of PSF by GSK3 controls CD45 alternative splicing. Mol. Cell 2010, 40, 126–137. [Google Scholar] [CrossRef]
- Ding, V.W.; Chen, R.H.; McCormick, F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J. Biol. Chem. 2000, 275, 32475–32481. [Google Scholar] [CrossRef] [PubMed]
- Tanji, C.; Yamamoto, H.; Yorioka, N.; Kohno, N.; Kikuchi, K.; Kikuchi, A. A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase- 3beta (GSK-3beta) and mediates protein kinase A-dependent inhibition of GSK-3beta. J. Biol. Chem. 2002, 277, 36955–36961. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Huang, J.; Buccafusca, R.; Garskof, J.; Valvezan, A.J.; Berry, G.T.; Klein, P.S. Glycogen synthase kinase-3 is essential for beta-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J. Clin. Investig. 2011, 121, 3756–3762. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Marion, S.; Rodriguiz, R.M.; Medvedev, I.O.; Sotnikova, T.D.; Ghisi, V.; Wetsel, W.C.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. A beta-arrestin 2 signaling complex mediates lithium action on behavior. Cell 2008, 132, 125–136. [Google Scholar] [CrossRef]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Niehrs, C. The role of Xenopus developmental biology in unraveling Wnt signalling and antero-posterior axis formation. Dev. Biol. 2022, 482, 1–6. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef] [PubMed]
- Wiese, K.E.; Nusse, R.; van Amerongen, R. Wnt signalling: Conquering complexity. Development 2018, 145, dev165902. [Google Scholar] [CrossRef] [PubMed]
- Sawa, H.; Korswagen, H.C. Wnt signaling in C. elegans. WormBook 2013, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.D.; Mullins, M.C. Cell signaling pathways controlling an axis organizing center in the zebrafish. Curr. Top. Dev. Biol. 2022, 150, 149–209. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Sharma, R.P. Wingless, a new mutant in Drosophila melanogaster. Dev. Biol. Inf. Sev. 1973, 50, 134. [Google Scholar]
- Morata, G.; Lawrence, P.A. The development of wingless, a homeotic mutation of Drosophila. Dev. Biol. 1977, 56, 227–240. [Google Scholar] [CrossRef]
- Baker, N.E. Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: The spatial distribution of a transcript in embryos. EMBO J. 1987, 6, 1765–1773. [Google Scholar] [CrossRef]
- Cabrera, C.V.; Alonso, M.C.; Johnston, P.; Phillips, R.G.; Lawrence, P.A. Phenocopies induced with antisense RNA identify the wingless gene. Cell 1987, 50, 659–663. [Google Scholar] [CrossRef]
- Perrimon, N.; Mahowald, A.P. Multiple functions of segment polarity genes in Drosophila. Dev. Biol. 1987, 119, 587–600. [Google Scholar] [CrossRef]
- Rijsewijk, F.; Schuermann, M.; Wagenaar, E.; Parren, P.; Weigel, D.; Nusse, R. The Drosophila Homolog of the Mouse Mammary Oncogene int-1 Is Identical to the Segment Polarity Gene wingless. Cell 1987, 50, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A new nomenclature for int-1 and related genes: The Wnt gene family [letter]. Cell 1991, 64, 231. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996, 272, 1023–1026. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Robbins, P.; El, G.M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.C.; Tonozuka, T.; Stamos, J.L.; Choi, H.J.; Weis, W.I. Mechanism of phosphorylation-dependent binding of APC to beta-catenin and its role in beta-catenin degradation. Mol. Cell 2004, 15, 511–521. [Google Scholar] [CrossRef]
- Liu, J.; Xing, Y.; Hinds, T.R.; Zheng, J.; Xu, W. The third 20 amino acid repeat is the tightest binding site of APC for beta-catenin. J. Mol. Biol. 2006, 360, 133–144. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Valvezan, A.J.; Zhang, F.; Diehl, J.A.; Klein, P.S. Adenomatous Polyposis Coli (APC) Regulates Multiple Signaling Pathways by Enhancing Glycogen Synthase Kinase-3 (GSK-3) Activity. J. Biol. Chem. 2012, 287, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- Hinoi, T.; Yamamoto, H.; Kishida, M.; Takada, S.; Kishida, S.; Kikuchi, A. Complex formation of adenomatous polyposis coli gene product and axin facilitates glycogen synthase kinase-3 beta-dependent phosphorylation of beta-catenin and down-regulates beta-catenin. J. Biol. Chem. 2000, 275, 34399–34406. [Google Scholar] [CrossRef]
- Tacchelly-Benites, O.; Wang, Z.; Yang, E.; Benchabane, H.; Tian, A.; Randall, M.P.; Ahmed, Y. Axin phosphorylation in both Wnt-off and Wnt-on states requires the tumor suppressor APC. PLoS Genet. 2018, 14, e1007178. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Wu, W.; Shen, J.; Bilic, J.; Fenger, U.; Stannek, P.; Glinka, A.; Niehrs, C. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature 2005, 438, 867–872. [Google Scholar] [CrossRef]
- Zeng, X.; Tamai, K.; Doble, B.; Li, S.; Huang, H.; Habas, R.; Okamura, H.; Woodgett, J.; He, X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 2005, 438, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.R.; Klein, A.M.; Kirschner, M.W. Kinetic responses of beta-catenin specify the sites of Wnt control. Science 2012, 338, 1337–1340. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.G.; Eldar-Finkelman, H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on beta-catenin in mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784. [Google Scholar] [CrossRef]
- Zhang, F.; Phiel, C.J.; Spece, L.; Gurvich, N.; Klein, P.S. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 2003, 278, 33067–33077. [Google Scholar] [CrossRef]
- Ji, L.; Lu, B.; Wang, Z.; Yang, Z.; Reece-Hoyes, J.; Russ, C.; Xu, W.; Cong, F. Identification of ICAT as an APC Inhibitor, Revealing Wnt-Dependent Inhibition of APC-Axin Interaction. Mol. Cell 2018, 72, 37–47.e34. [Google Scholar] [CrossRef]
- Tran, H.; Polakis, P. Reversible modification of adenomatous polyposis coli (APC) with K63-linked polyubiquitin regulates the assembly and activity of the beta-catenin destruction complex. J. Biol. Chem. 2012, 287, 28552–28563. [Google Scholar] [CrossRef]
- Mukherjee, A.; Dhar, N.; Stathos, M.; Schaffer, D.V.; Kane, R.S. Understanding How Wnt Influences Destruction Complex Activity and beta-Catenin Dynamics. iScience 2018, 6, 13–21. [Google Scholar] [CrossRef]
- Lybrand, D.B.; Naiman, M.; Laumann, J.M.; Boardman, M.; Petshow, S.; Hansen, K.; Scott, G.; Wehrli, M. Destruction complex dynamics: Wnt/beta-catenin signaling alters Axin-GSK3beta interactions in vivo. Development 2019, 146, dev164145. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, J.P.; Emmink, B.L.; Nojima, H.; Kranenburg, O.; Maurice, M.M. Wnt signalling induces accumulation of phosphorylated beta-catenin in two distinct cytosolic complexes. Open Biol. 2014, 4, 140120. [Google Scholar] [CrossRef] [PubMed]
- Valvezan, A.J.; Huang, J.; Lengner, C.J.; Pack, M.; Klein, P.S. Oncogenic mutations in adenomatous polyposis coli (Apc) activate mechanistic target of rapamycin complex 1 (mTORC1) in mice and zebrafish. Dis. Models Mech. 2014, 7, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Lee, S.H.; Kim, H.; Yum, S.; Stamos, J.L.; Xu, Y.; Lee, S.J.; Lee, J.; Oh, S.; Han, J.K.; et al. Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta-catenin signaling. PLoS ONE 2008, 3, e4046. [Google Scholar] [CrossRef]
- Stamos, J.L.; Chu, M.L.; Enos, M.D.; Shah, N.; Weis, W.I. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. eLife 2014, 3, e01998. [Google Scholar] [CrossRef]
- Wu, G.; Huang, H.; Garcia Abreu, J.; He, X. Inhibition of GSK3 phosphorylation of beta-catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS ONE 2009, 4, e4926. [Google Scholar] [CrossRef]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef]
- McManus, E.J.; Sakamoto, K.; Armit, L.J.; Ronaldson, L.; Shpiro, N.; Marquez, R.; Alessi, D.R. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005, 24, 1571–1583. [Google Scholar] [CrossRef]
- Berra, Y. The Yogi Book: I Really Didn’t Say All the Things I Said; Workman Publishing Company: New York, NY, USA, 1999. [Google Scholar]
- Kim, N.G.; Xu, C.; Gumbiner, B.M. Identification of targets of the Wnt pathway destruction complex in addition to beta-catenin. Proc. Natl. Acad. Sci. USA 2009, 106, 5165–5170. [Google Scholar] [CrossRef]
- Xu, C.; Kim, N.G.; Gumbiner, B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Eisinger, A.L.; Nadauld, L.D.; Shelton, D.N.; Peterson, P.W.; Phelps, R.A.; Chidester, S.; Stafforini, D.M.; Prescott, S.M.; Jones, D.A. The adenomatous polyposis coli tumor suppressor gene regulates expression of cyclooxygenase-2 by a mechanism that involves retinoic acid. J. Biol. Chem. 2006, 281, 20474–20482. [Google Scholar] [CrossRef] [PubMed]
- Phelps, R.A.; Chidester, S.; Dehghanizadeh, S.; Phelps, J.; Sandoval, I.T.; Rai, K.; Broadbent, T.; Sarkar, S.; Burt, R.W.; Jones, D.A. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 2009, 137, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Karaulanov, E.; Berger, B.S.; Huang, Y.L.; Niehrs, C. Mitotic wnt signaling promotes protein stabilization and regulates cell size. Mol. Cell 2014, 54, 663–674. [Google Scholar] [CrossRef]
- Schlesinger, A.; Shelton, C.A.; Maloof, J.N.; Meneghini, M.; Bowerman, B. Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev. 1999, 13, 2028–2038. [Google Scholar] [CrossRef]
- Rocheleau, C.E.; Downs, W.D.; Lin, R.; Wittmann, C.; Bei, Y.; Cha, Y.H.; Ali, M.; Priess, J.R.; Mello, C.C. Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos. Cell 1997, 90, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, K.; Mizumoto, K.; Sawa, H. Wnt regulates spindle asymmetry to generate asymmetric nuclear beta-catenin in C. elegans. Cell 2011, 146, 942–954. [Google Scholar] [CrossRef]
- Caldwell, C.M.; Kaplan, K.B. The role of APC in mitosis and in chromosome instability. Adv. Exp. Med. Biol. 2009, 656, 51–64. [Google Scholar]
- Hanson, C.A.; Miller, J.R. Non-traditional roles for the Adenomatous Polyposis Coli (APC) tumor suppressor protein. Gene 2005, 361, 1–12. [Google Scholar] [CrossRef]
- Wakefield, J.G.; Stephens, D.J.; Tavare, J.M. A role for glycogen synthase kinase-3 in mitotic spindle dynamics and chromosome alignment. J. Cell Sci. 2003, 116, 637–646. [Google Scholar] [CrossRef]
- Cai, J.; Maitra, A.; Anders, R.A.; Taketo, M.M.; Pan, D. beta-Catenin destruction complex-independent regulation of Hippo-YAP signaling by APC in intestinal tumorigenesis. Genes Dev. 2015, 29, 1493–1506. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Nadauld, L.D.; Sandoval, I.T.; Chidester, S.; Yost, H.J.; Jones, D.A. Adenomatous polyposis coli control of retinoic acid biosynthesis is critical for zebrafish intestinal development and differentiation. J. Biol. Chem. 2004, 279, 51581–51589. [Google Scholar] [CrossRef] [PubMed]
- Nadauld, L.D.; Phelps, R.; Moore, B.C.; Eisinger, A.; Sandoval, I.T.; Chidester, S.; Peterson, P.W.; Manos, E.J.; Sklow, B.; Burt, R.W.; et al. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J. Biol. Chem. 2006, 281, 37828–37835. [Google Scholar] [CrossRef]
- Fukuda, T.; Kokabu, S.; Ohte, S.; Sasanuma, H.; Kanomata, K.; Yoneyama, K.; Kato, H.; Akita, M.; Oda, H.; Katagiri, T. Canonical Wnts and BMPs cooperatively induce osteoblastic differentiation through a GSK3beta-dependent and beta-catenin-independent mechanism. Differentiation 2010, 80, 46–52. [Google Scholar] [CrossRef]
- Miclea, R.L.; van der Horst, G.; Robanus-Maandag, E.C.; Löwik, C.W.; Oostdijk, W.; Wit, J.M.; Karperien, M. Apc bridges Wnt/β-catenin and BMP signaling during osteoblast differentiation of KS483 cells. Exp. Cell Res. 2011, 317, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, L.C.; Eivers, E.; Ikeda, A.; Hurtado, C.; Kuroda, H.; Pera, E.M.; De Robertis, E.M. Integrating patterning signals: Wnt/GSK3 regulates the duration of the BMP/Smad1 signal. Cell 2007, 131, 980–993. [Google Scholar] [CrossRef]
- Park, K.S.; Jeon, S.H.; Kim, S.E.; Bahk, Y.Y.; Holmen, S.L.; Williams, B.O.; Chung, K.C.; Surh, Y.J.; Choi, K.Y. APC inhibits ERK pathway activation and cellular proliferation induced by RAS. J. Cell Sci. 2006, 119, 819–827. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y.; Wang, X.; Evers, B.M. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene 2006, 25, 43–50. [Google Scholar] [CrossRef]
- Zhai, P.; Gao, S.; Holle, E.; Yu, X.; Yatani, A.; Wagner, T.; Sadoshima, J. Glycogen synthase kinase-3alpha reduces cardiac growth and pressure overload-induced cardiac hypertrophy by inhibition of extracellular signal-regulated kinases. J. Biol. Chem. 2007, 282, 33181–33191. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, Y.; Bersenev, A.; O’Brien, W.T.; Tong, W.; Emerson, S.G.; Klein, P.S. Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J. Clin. Investig. 2009, 119, 3519–3529. [Google Scholar] [CrossRef]
- Qian, Z.; Chen, L.; Fernald, A.A.; Williams, B.O.; Le Beau, M.M. A critical role for Apc in hematopoietic stem and progenitor cell survival. J. Exp. Med. 2008, 205, 2163–2175. [Google Scholar] [CrossRef]
- Guezguez, B.; Almakadi, M.; Benoit, Y.D.; Shapovalova, Z.; Rahmig, S.; Fiebig-Comyn, A.; Casado, F.L.; Tanasijevic, B.; Bresolin, S.; Masetti, R.; et al. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of Human Acute Leukemia. Cancer Cell 2016, 29, 61–74. [Google Scholar] [CrossRef]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Fearon, E.R.; Cho, K.R.; Nigro, J.M.; Kern, S.E.; Simons, J.W.; Ruppert, J.M.; Hamilton, S.R.; Preisinger, A.C.; Thomas, G.; Kinzler, K.W.; et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science 1990, 247, 49–56. [Google Scholar] [CrossRef]
- Abbott, J.; Nathke, I.S. The adenomatous polyposis coli protein 30 years on. Semin. Cell Dev. Biol. 2023, 150–151, 28–34. [Google Scholar] [CrossRef]
- Dow, L.E.; O’Rourke, K.P.; Simon, J.; Tschaharganeh, D.F.; van Es, J.H.; Clevers, H.; Lowe, S.W. Apc Restoration Promotes Cellular Differentiation and Reestablishes Crypt Homeostasis in Colorectal Cancer. Cell 2015, 161, 1539–1552. [Google Scholar] [CrossRef]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Fenton, S.E.; Burns, M.C.; Kalyan, A. Epidemiology, mutational landscape and staging of hepatocellular carcinoma. Chin. Clin. Oncol. 2021, 10, 2. [Google Scholar] [CrossRef]
- Groenewald, W.; Lund, A.H.; Gay, D.M. The Role of WNT Pathway Mutations in Cancer Development and an Overview of Therapeutic Options. Cells 2023, 12, 990. [Google Scholar] [CrossRef] [PubMed]
- Park, W.J.; Kim, M.J. A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells 2023, 12, 1110. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; Smith, D.I.; et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef] [PubMed]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Dai, Q.; Sun, D.F.; Xiong, H.; Tian, X.Q.; Gao, F.H.; Xu, M.H.; Chen, G.Q.; Han, Z.G.; Fang, J.Y. mTOR signaling pathway is a target for the treatment of colorectal cancer. Ann. Surg. Oncol. 2009, 16, 2617–2628. [Google Scholar] [CrossRef]
- Nozawa, H.; Watanabe, T.; Nagawa, H. Phosphorylation of ribosomal p70 S6 kinase and rapamycin sensitivity in human colorectal cancer. Cancer Lett. 2007, 251, 105–113. [Google Scholar] [CrossRef]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27 (Suppl. 2), S43–S51. [Google Scholar] [CrossRef]
- Metcalfe, C.; Ibrahim, A.E.; Graeb, M.; de la Roche, M.; Schwarz-Romond, T.; Fiedler, M.; Winton, D.J.; Corfield, A.; Bienz, M. Dvl2 promotes intestinal length and neoplasia in the ApcMin mouse model for colorectal cancer. Cancer Res. 2010, 70, 6629–6638. [Google Scholar] [CrossRef]
- Fujishita, T.; Aoki, K.; Lane, H.A.; Aoki, M.; Taketo, M.M. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proc. Natl. Acad. Sci. USA 2008, 105, 13544–13549. [Google Scholar] [CrossRef]
- Parihar, M.; Dodds, S.G.; Hubbard, G.; Javors, M.A.; Strong, R.; Hasty, P.; Sharp, Z.D. Rapamycin Extends Life Span in Apc(Min/+) Colon Cancer FAP Model. Clin. Color. Cancer 2021, 20, e61–e70. [Google Scholar] [CrossRef]
- Brandt, M.; Grazioso, T.P.; Fawal, M.A.; Tummala, K.S.; Torres-Ruiz, R.; Rodriguez-Perales, S.; Perna, C.; Djouder, N. mTORC1 Inactivation Promotes Colitis-Induced Colorectal Cancer but Protects from APC Loss-Dependent Tumorigenesis. Cell Metab. 2018, 27, 118–135.e118. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, N.; Yousefi, M.; Nakauka-Ddamba, A.; Li, F.; Parada, K.; Rao, S.; Minuesa, G.; Katz, Y.; Gregory, B.D.; et al. Transformation of the intestinal epithelium by the MSI2 RNA-binding protein. Nat. Commun. 2015, 6, 6517. [Google Scholar] [CrossRef]
- Hasty, P.; Livi, C.B.; Dodds, S.G.; Jones, D.; Strong, R.; Javors, M.; Fischer, K.E.; Sloane, L.; Murthy, K.; Hubbard, G.; et al. eRapa restores a normal life span in a FAP mouse model. Cancer Prev. Res. 2014, 7, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef]
- Koehl, G.E.; Spitzner, M.; Ousingsawat, J.; Schreiber, R.; Geissler, E.K.; Kunzelmann, K. Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APC(Min/+) mice. Oncogene 2010, 29, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Fricke, S.L.; Payne, S.N.; Favreau, P.F.; Kratz, J.D.; Pasch, C.A.; Foley, T.M.; Yueh, A.E.; Van De Hey, D.R.; Depke, M.G.; Korkos, D.P.; et al. MTORC1/2 Inhibition as a Therapeutic Strategy for PIK3CA Mutant Cancers. Mol. Cancer Ther. 2019, 18, 346–355. [Google Scholar] [CrossRef]
- Haramis, A.P.; Hurlstone, A.; van der Velden, Y.; Begthel, H.; van den Born, M.; Offerhaus, G.J.; Clevers, H.C. Adenomatous polyposis coli-deficient zebrafish are susceptible to digestive tract neoplasia. EMBO Rep. 2006, 7, 444–449. [Google Scholar] [CrossRef]
- Hurlstone, A.F.; Haramis, A.P.; Wienholds, E.; Begthel, H.; Korving, J.; Van Eeden, F.; Cuppen, E.; Zivkovic, D.; Plasterk, R.H.; Clevers, H. The Wnt/beta-catenin pathway regulates cardiac valve formation. Nature 2003, 425, 633–637. [Google Scholar] [CrossRef]
- Goessling, W.; North, T.E.; Lord, A.M.; Ceol, C.; Lee, S.; Weidinger, G.; Bourque, C.; Strijbosch, R.; Haramis, A.P.; Puder, M.; et al. APC mutant zebrafish uncover a changing temporal requirement for wnt signaling in liver development. Dev. Biol. 2008, 320, 161–174. [Google Scholar] [CrossRef]
- Foley, P.J.; Scheri, R.P.; Smolock, C.J.; Pippin, J.; Green, D.W.; Drebin, J.A. Targeted suppression of beta-catenin blocks intestinal adenoma formation in APC Min mice. J. Gastrointest. Surg. 2008, 12, 1452–1458. [Google Scholar] [CrossRef]
- Scholer-Dahirel, A.; Schlabach, M.R.; Loo, A.; Bagdasarian, L.; Meyer, R.; Guo, R.; Woolfenden, S.; Yu, K.K.; Markovits, J.; Killary, K.; et al. Maintenance of adenomatous polyposis coli (APC)-mutant colorectal cancer is dependent on Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 17135–17140. [Google Scholar] [CrossRef] [PubMed]
- Romagnolo, B.; Berrebi, D.; Saadi-Keddoucci, S.; Porteu, A.; Pichard, A.L.; Peuchmaur, M.; Vandewalle, A.; Kahn, A.; Perret, C. Intestinal dysplasia and adenoma in transgenic mice after overexpression of an activated beta-catenin. Cancer Res. 1999, 59, 3875–3879. [Google Scholar] [PubMed]
- Harada, N.; Tamai, Y.; Ishikawa, T.; Sauer, B.; Takaku, K.; Oshima, M.; Taketo, M.M. Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J. 1999, 18, 5931–5942. [Google Scholar] [CrossRef] [PubMed]
- Amos-Landgraf, J.M.; Kwong, L.N.; Kendziorski, C.M.; Reichelderfer, M.; Torrealba, J.; Weichert, J.; Haag, J.D.; Chen, K.S.; Waller, J.L.; Gould, M.N.; et al. A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 4036–4041. [Google Scholar] [CrossRef]
- Anderson, C.B.; Neufeld, K.L.; White, R.L. Subcellular distribution of Wnt pathway proteins in normal and neoplastic colon. Proc. Natl. Acad. Sci. USA 2002, 99, 8683–8688. [Google Scholar] [CrossRef]
- Blaker, H.; Sutter, C.; Kadmon, M.; Otto, H.F.; Von Knebel-Doeberitz, M.; Gebert, J.; Helmke, B.M. Analysis of somatic APC mutations in rare extracolonic tumors of patients with familial adenomatous polyposis coli. Genes Chromosomes Cancer 2004, 41, 93–98. [Google Scholar] [CrossRef]
- Kobayashi, M.; Honma, T.; Matsuda, Y.; Suzuki, Y.; Narisawa, R.; Ajioka, Y.; Asakura, H. Nuclear translocation of beta-catenin in colorectal cancer. Br. J. Cancer 2000, 82, 1689–1693. [Google Scholar] [CrossRef]
- Phelps, R.A.; Broadbent, T.J.; Stafforini, D.M.; Jones, D.A. New perspectives on APC control of cell fate and proliferation in colorectal cancer. Cell Cycle 2009, 8, 2549–2556. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Hurcombe, J.A.; Hartley, P.; Lay, A.C.; Ni, L.; Bedford, J.J.; Leader, J.P.; Singh, S.; Murphy, A.; Scudamore, C.L.; Marquez, E.; et al. Podocyte GSK3 is an evolutionarily conserved critical regulator of kidney function. Nat. Commun. 2019, 10, 403. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Konsavage, W.M., Jr.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [PubMed]
- Azzolin, L.; Zanconato, F.; Bresolin, S.; Forcato, M.; Basso, G.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Role of TAZ as mediator of Wnt signaling. Cell 2012, 151, 1443–1456. [Google Scholar] [CrossRef]
- Gregorieff, A.; Liu, Y.; Inanlou, M.R.; Khomchuk, Y.; Wrana, J.L. Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 2015, 526, 715–718. [Google Scholar] [CrossRef]
- Cheung, P.; Xiol, J.; Dill, M.T.; Yuan, W.C.; Panero, R.; Roper, J.; Osorio, F.G.; Maglic, D.; Li, Q.; Gurung, B.; et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell 2020, 27, 590–604.e599. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, L.; Westman, J.; Hallgren, J.; Osby, U.; Backlund, L. Lithium treatment and cancer incidence in bipolar disorder. Bipolar Disord. 2016, 18, 33–40. [Google Scholar] [CrossRef]
- Ozerdem, A.; Ceylan, D.; Targitay, B. The Relationship Between Lithium and Cancer Proliferation: A Case-Based Review of the Literature. Curr. Drug Metab. 2018, 19, 653–662. [Google Scholar] [CrossRef]
- Huang, R.Y.; Hsieh, K.P.; Huang, W.W.; Yang, Y.H. Use of lithium and cancer risk in patients with bipolar disorder: Population-based cohort study. Br. J. Psychiatry 2016, 209, 393–399. [Google Scholar] [CrossRef]
- Linssen, J.D.G.; van Neerven, S.M.; Aelvoet, A.S.; Elbers, C.C.; Vermeulen, L.; Dekker, E. The CHAMP-study: The CHemopreventive effect of lithium in familial AdenoMatous Polyposis; study protocol of a phase II trial. BMC Gastroenterol. 2022, 22, 383. [Google Scholar] [CrossRef]
- Harrington, C.T.; Sotillo, E.; Robert, A.; Hayer, K.E.; Bogusz, A.M.; Psathas, J.; Yu, D.; Taylor, D.; Dang, C.V.; Klein, P.S.; et al. Transient stabilization, rather than inhibition, of MYC amplifies extrinsic apoptosis and therapeutic responses in refractory B-cell lymphoma. Leukemia 2019, 33, 2429–2441. [Google Scholar] [CrossRef]
- Kim, W.Y.; Wang, X.; Wu, Y.H.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Furth, E.E.; Rustgi, A.K.; Klein, P.S. When You Come to a Fork in the Road, Take It: Wnt Signaling Activates Multiple Pathways through the APC/Axin/GSK-3 Complex. Cells 2023, 12, 2256. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12182256
Li C, Furth EE, Rustgi AK, Klein PS. When You Come to a Fork in the Road, Take It: Wnt Signaling Activates Multiple Pathways through the APC/Axin/GSK-3 Complex. Cells. 2023; 12(18):2256. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12182256
Chicago/Turabian StyleLi, Chenchen, Emma E. Furth, Anil K. Rustgi, and Peter S. Klein. 2023. "When You Come to a Fork in the Road, Take It: Wnt Signaling Activates Multiple Pathways through the APC/Axin/GSK-3 Complex" Cells 12, no. 18: 2256. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12182256