

Okadaic Acid Activates JAK/STAT Signaling to Affect Xenobiotic Metabolism in HepaRG Cells
 , , , and
, , , and 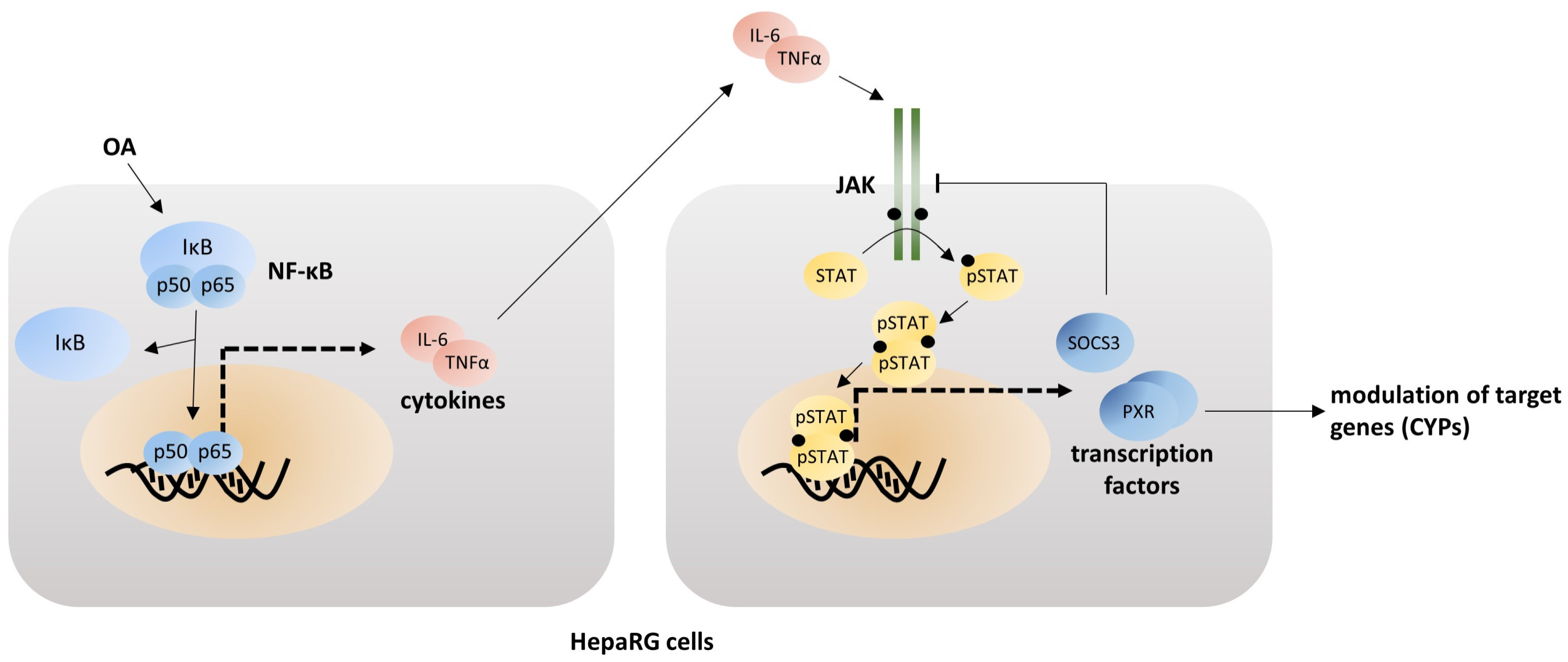{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Cultivation
2.3. Gene Expression Analysis
2.4. Western Blotting
2.5. Confocal Microscopy
2.6. Protein Quantification
2.7. PXR and RXRα Transactivation Assay
2.8. Statistical Analysis
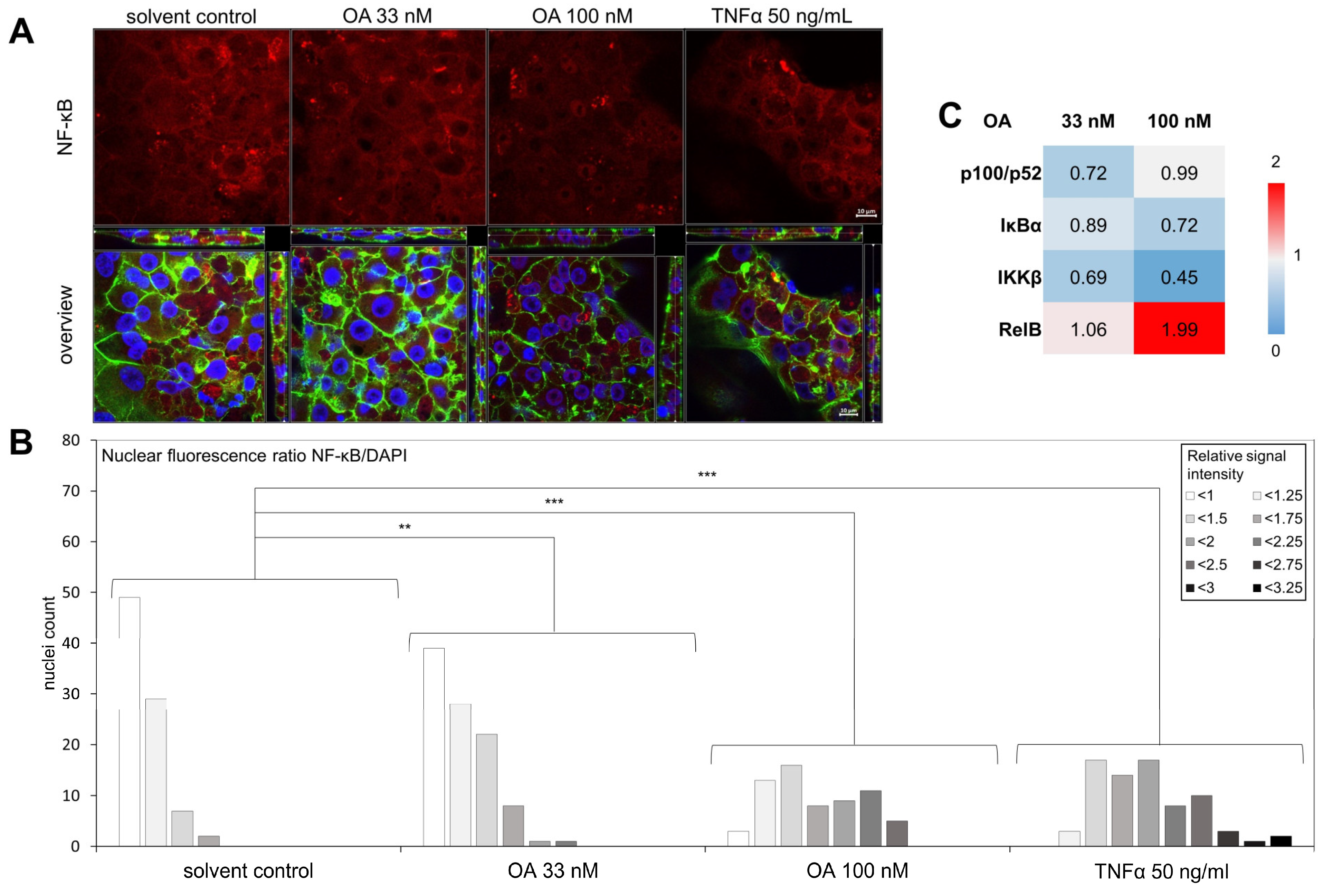
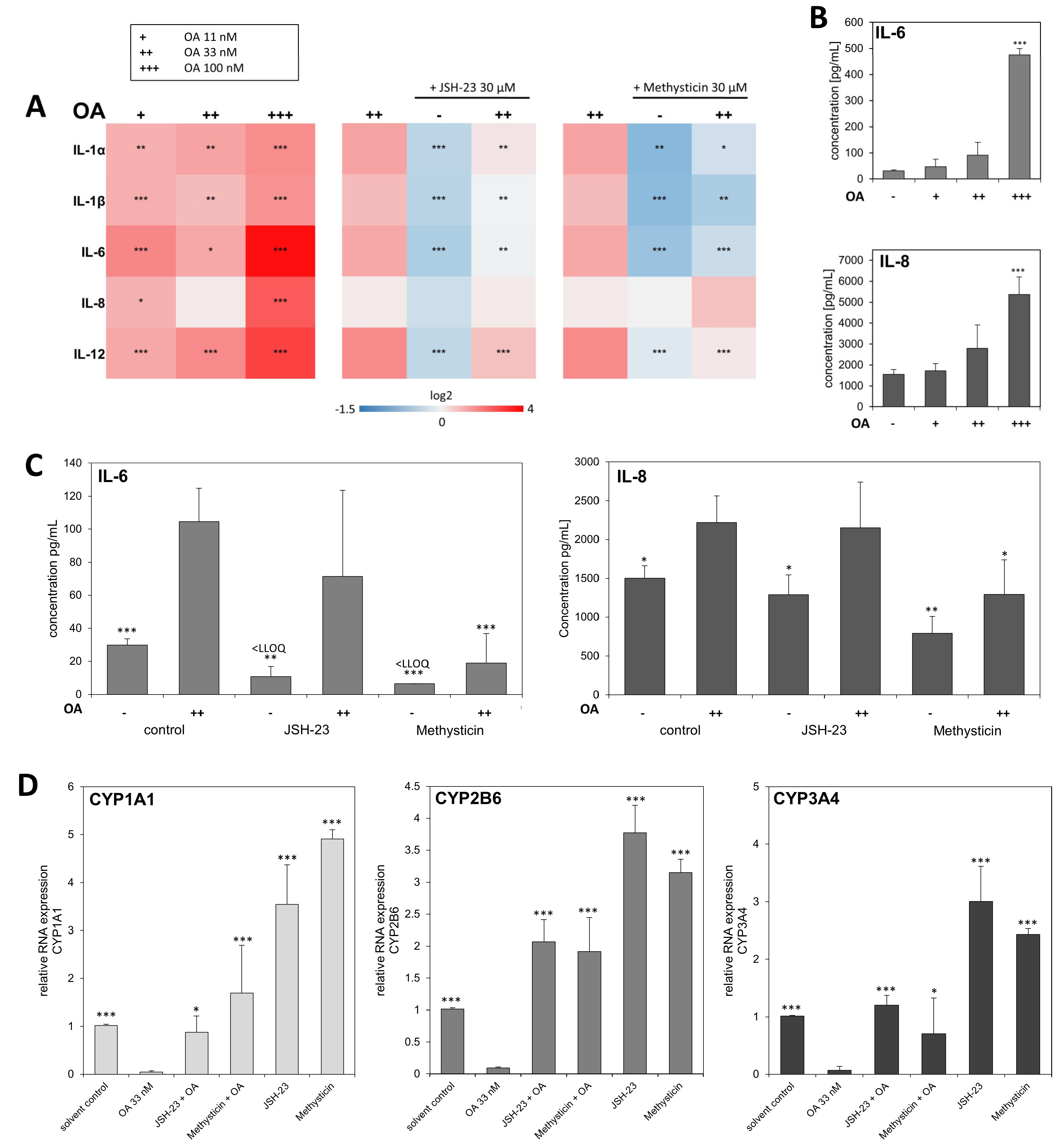
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EFSA. Scientific Opinion of the Panel on Contaminants in the Food chain on a request from the European Commission on marine biotoxins in shellfish—Okadaic acid and analogue. EFSA J. 2008, 589, 1–12. [Google Scholar]
- Van Dolah, F.M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 2000, 108 (Suppl. 1), 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO. Marine biotoxins. FAO Food Nutr. Pap. 2004, 80, 53–92. [Google Scholar]
- Ferron, P.J.; Hogeveen, K.; Fessard, V.; Le Hégarat, L. Comparative analysis of the cytotoxic effects of okadaic acid-group toxins on human intestinal cell lines. Mar. Drugs 2014, 12, 4616–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fessard, V.; Grosse, Y.; Pfohl-Leszkowicz, A.; Puiseux-Dao, S. Okadaic acid treatment induces DNA adduct formation in BHK21 C13 fibroblasts and HESV keratinocytes. Mutat. Res. 1996, 361, 133–141. [Google Scholar] [CrossRef]
- Le Hégarat, L.; Jacquin, A.G.; Bazin, E.; Fessard, V. Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ. Toxicol. 2006, 21, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, A.; Stempin, S.; Al-Hamwi, R.; Lampen, A. Embryotoxic effects of the marine biotoxin okadaic acid on murine embryonic stem cells. Toxicon 2010, 55, 855–863. [Google Scholar] [CrossRef]
- Ariu, F.; Fois, S.; Bebbere, D.; Ledda, S.; Rosati, I.; Zedda, M.T.; Pau, S.; Bogliolo, L. The effect of okadaic acid on meiotic maturation of canine oocytes of different size. Theriogenology 2012, 77, 46–52. [Google Scholar] [CrossRef]
- Matias, W.G.; Creppy, E.E. Transplacental passage of [3H]-okadaic acid in pregnant mice measured by radioactivity and high-performance liquid chromatography. Hum. Exp. Toxicol. 1996, 15, 226–230. [Google Scholar] [CrossRef]
- Jiménez-Cárcamo, D.; García, C.; Contreras, H.R. Toxins of Okadaic Acid-Group Increase Malignant Properties in Cells of Colon Cancer. Toxins 2020, 12, 179. [Google Scholar] [CrossRef] [Green Version]
- Messner, D.J.; Romeo, C.; Boynton, A.; Rossie, S. Inhibition of PP2A, but not PP5, mediates p53 activation by low levels of okadaic acid in rat liver epithelial cells. J. Cell. Biochem. 2006, 99, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Cordier, S.; Monfort, C.; Miossec, L.; Richardson, S.; Belin, C. Ecological Analysis of Digestive Cancer Mortality Related to Contamination by Diarrhetic Shellfish Poisoning Toxins along the Coasts of France. Environ. Res. 2000, 84, 145–150. [Google Scholar] [CrossRef]
- Lopez-Rodas, V.; Maneiro, E.; Martinez, J.; Navarro, M.; Costas, E. Harmful algal blooms, red tides and human health: Diarrhetic shellfish poisoning and colorectal cancer. An. R. Acad. Farm. 2006, 72, 391–408. [Google Scholar]
- Manerio, E.; Rodas, V.L.; Costas, E.; Hernandez, J.M. Shellfish consumption: A major risk factor for colorectal cancer. Med. Hypotheses 2008, 70, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar] [CrossRef]
- Opsahl, J.A.; Ljostveit, S.; Solstad, T.; Risa, K.; Roepstorff, P.; Fladmark, K.E. Identification of dynamic changes in proteins associated with the cellular cytoskeleton after exposure to okadaic acid. Mar. Drugs 2013, 11, 1763–1782. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Lin, L.; Wang, D.-Z. Quantitative proteomic analysis reveals novel insights into hepatic toxicity in mice exposed chronically to okadaic acid. Sci. Total Environ. 2021, 775, 145772. [Google Scholar] [CrossRef]
- Dietrich, J.; Grass, I.; Günzel, D.; Herek, S.; Braeuning, A.; Lampen, A.; Hessel-Pras, S. The marine biotoxin okadaic acid affects intestinal tight junction proteins in human intestinal cells. Toxicol. Vitr. 2019, 58, 150–160. [Google Scholar] [CrossRef]
- Wuerger, L.T.D.; Hammer, H.S.; Hofmann, U.; Kudiabor, F.; Sieg, H.; Braeuning, A. Okadaic acid influences xenobiotic metabolism in HepaRG cells. EXCLI J. 2022, 21, 1053–1065. [Google Scholar]
- Thévenin, C.; Kim, S.J.; Rieckmann, P.; Fujiki, H.; Norcross, M.A.; Sporn, M.B.; Fauci, A.S.; Kehrl, J.H. Induction of nuclear factor-kappa B and the human immunodeficiency virus long terminal repeat by okadaic acid, a specific inhibitor of phosphatases 1 and 2A. New Biol. 1990, 2, 793–800. [Google Scholar] [PubMed]
- Ozaki, A.; Morimoto, H.; Tanaka, H.; Okamura, H.; Yoshida, K.; Amorim, B.R.; Haneji, T. Okadaic acid induces phosphorylation of p65NF-kappaB on serine 536 and activates NF-kappaB transcriptional activity in human osteoblastic MG63 cells. J. Cell. Biochem. 2006, 99, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Ohmori, Y.; Chang, P.L. Production of chemokine CXCL1/KC by okadaic acid through the nuclear factor-kappaB pathway. Carcinogenesis 2006, 27, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Goto, K.; Yoshida, K.; Okamura, H.; Morimoto, H.; Kito, S.; Fukuda, J.; Haneji, T. Okadaic acid stimulates expression of Fas receptor and Fas ligand by activation of nuclear factor kappa-B in human oral squamous carcinoma cells. Oral Oncol. 2004, 40, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Miskolci, V.; Castro-Alcaraz, S.; Nguyen, P.; Vancura, A.; Davidson, D.; Vancurova, I. Okadaic acid induces sustained activation of NFkappaB and degradation of the nuclear IkappaBalpha in human neutrophils. Arch. Biochem. Biophys. 2003, 417, 44–52. [Google Scholar] [CrossRef]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef]
- Keller, R.; Klein, M.; Thomas, M.; Dräger, A.; Metzger, U.; Templin, M.F.; Joos, T.O.; Thasler, W.E.; Zell, A.; Zanger, U.M. Coordinating Role of RXRα in Downregulating Hepatic Detoxification during Inflammation Revealed by Fuzzy-Logic Modeling. PLoS Comput. Biol. 2016, 12, e1004431. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, H.; Wang, P.; Wang, J.; Zou, L. The roles of SOCS3 and STAT3 in bacterial infection and inflammatory diseases. Scand. J. Immunol. 2018, 88, e12727. [Google Scholar] [CrossRef] [Green Version]
- Kanebratt, K.P.; Andersson, T.B. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab. Dispos. 2008, 36, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Tascher, G.; Burban, A.; Camus, S.; Plumel, M.; Chanon, S.; Le Guevel, R.; Shevchenko, V.; Van Dorsselaer, A.; Lefai, E.; Guguen-Guillouzo, C.; et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells 2019, 8, 192. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Mueller, D.; Schevchenko, V.; Noor, F. Long-term maintenance of HepaRG cells in serum-free conditions and application in a repeated dose study. J. Appl. Toxicol. 2014, 34, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Sieg, H.; Ellermann, A.L.; Maria Kunz, B.; Jalili, P.; Burel, A.; Hogeveen, K.; Böhmert, L.; Chevance, S.; Braeuning, A.; Gauffre, F.; et al. Aluminum in liver cells—The element species matters. Nanotoxicology 2019, 13, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Treindl, F.; Ruprecht, B.; Beiter, Y.; Schultz, S.; Döttinger, A.; Staebler, A.; Joos, T.O.; Kling, S.; Poetz, O.; Fehm, T.; et al. A bead-based western for high-throughput cellular signal transduction analyses. Nat. Commun. 2016, 7, 12852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckert, C.; Hessel, S.; Lampen, A.; Braeuning, A. Utility of an appropriate reporter assay: Heliotrine interferes with GAL4/upstream activation sequence-driven reporter gene systems. Anal. Biochem. 2015, 487, 45–48. [Google Scholar] [CrossRef]
- Hampf, M.; Gossen, M. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006, 356, 94–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [Green Version]
- Hin Tang, J.J.; Hao Thng, D.K.; Lim, J.J.; Toh, T.B. JAK/STAT signaling in hepatocellular carcinoma. Hepat. Oncol. 2020, 7, Hep18. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.G.M.; Ghosh, A.; Variya, B.; Santharam, M.A.; Kandhi, R.; Ramanathan, S.; Ilangumaran, S. Hepatocyte growth control by SOCS1 and SOCS3. Cytokine 2019, 121, 154733. [Google Scholar] [CrossRef]
- Tanner, N.; Kubik, L.; Luckert, C.; Thomas, M.; Hofmann, U.; Zanger, U.M.; Böhmert, L.; Lampen, A.; Braeuning, A. Regulation of Drug Metabolism by the Interplay of Inflammatory Signaling, Steatosis, and Xeno-Sensing Receptors in HepaRG Cells. Drug Metab. Dispos. 2018, 46, 326–335. [Google Scholar] [CrossRef] [Green Version]
- DiDonato, J.A.; Hayakawa, M.; Rothwarf, D.M.; Zandi, E.; Karin, M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 1997, 388, 548–554. [Google Scholar] [CrossRef]
- Li, S.; Wang, L.; Berman, M.A.; Zhang, Y.; Dorf, M.E. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol. Cell 2006, 24, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Osaki, K.; Kanamoto, M.; Nakao, Y.; Takahashi, E.; Higuchi, T.; Kamata, H. Distinct B subunits of PP2A regulate the NF-κB signalling pathway through dephosphorylation of IKKβ, IκBα and RelA. FEBS Lett. 2017, 591, 4083–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.M.; Kim, M.H.; Kim, B.H.; Jung, S.H.; Kim, Y.S.; Park, H.J.; Hong, J.T.; Min, K.R.; Kim, Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004, 571, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Shaik, A.A.; Hermanson, D.L.; Xing, C. Identification of methysticin as a potent and non-toxic NF-kappaB inhibitor from kava, potentially responsible for kava’s chemopreventive activity. Bioorg. Med. Chem. Lett. 2009, 19, 5732–5736. [Google Scholar] [CrossRef] [Green Version]
- Folmer, F.; Blasius, R.; Morceau, F.; Tabudravu, J.; Dicato, M.; Jaspars, M.; Diederich, M. Inhibition of TNFalpha-induced activation of nuclear factor kappaB by kava (Piper methysticum) derivatives. Biochem. Pharmacol. 2006, 71, 1206–1218. [Google Scholar] [CrossRef]
- Klein, M.; Thomas, M.; Hofmann, U.; Seehofer, D.; Damm, G.; Zanger, U.M. A systematic comparison of the impact of inflammatory signaling on absorption, distribution, metabolism, and excretion gene expression and activity in primary human hepatocytes and HepaRG cells. Drug Metab. Dispos. 2015, 43, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Staudinger, J.L. Repression of PXR-mediated induction of hepatic CYP3A gene expression by protein kinase C. Biochem. Pharmacol. 2005, 69, 867–873. [Google Scholar] [CrossRef]
- Lv, C.; Huang, L. Xenobiotic receptors in mediating the effect of sepsis on drug metabolism. Acta Pharm. Sin. B 2020, 10, 33–41. [Google Scholar] [CrossRef]
- Moreau, A.; Vilarem, M.J.; Maurel, P.; Pascussi, J.M. Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response. Mol. Pharm. 2008, 5, 35–41. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mackowiak, B.; Wang, H. Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim. Biophys. Acta 2016, 1859, 1130–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wuerger, L.T.D.; Kudiabor, F.; Alarcan, J.; Templin, M.; Poetz, O.; Sieg, H.; Braeuning, A. Okadaic Acid Activates JAK/STAT Signaling to Affect Xenobiotic Metabolism in HepaRG Cells. Cells 2023, 12, 770. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12050770
Wuerger LTD, Kudiabor F, Alarcan J, Templin M, Poetz O, Sieg H, Braeuning A. Okadaic Acid Activates JAK/STAT Signaling to Affect Xenobiotic Metabolism in HepaRG Cells. Cells. 2023; 12(5):770. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12050770
Chicago/Turabian StyleWuerger, Leonie T. D., Felicia Kudiabor, Jimmy Alarcan, Markus Templin, Oliver Poetz, Holger Sieg, and Albert Braeuning. 2023. "Okadaic Acid Activates JAK/STAT Signaling to Affect Xenobiotic Metabolism in HepaRG Cells" Cells 12, no. 5: 770. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12050770