
Plasma Membrane Blebbing Is Controlled by Subcellular Distribution of Vimentin Intermediate Filaments
 , ,
, ,
Abstract
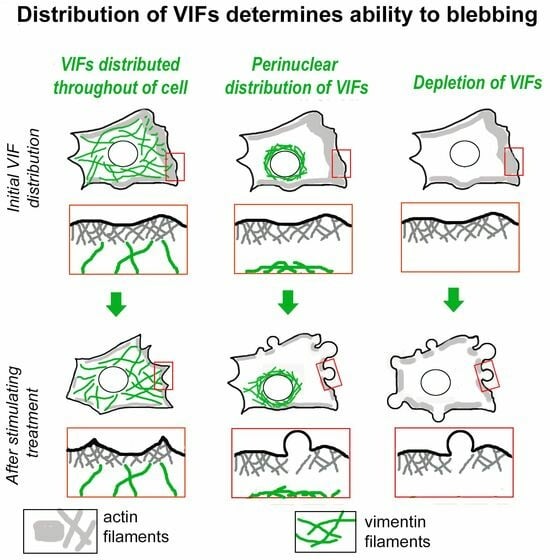
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Antibodies and Reagents
2.3. Western Blot Analysis
2.4. Light Microscopy
2.5. Correlative Platinum Replica Electron Microscopy (PREM)
2.6. Image Analysis
2.7. Statistical Analysis
3. Results
3.1. Distribution of Intermediate Filaments Differentiates Cells That Exhibit or Do Not Exhibit Induced Blebbing
3.2. Subcellular Distribution of VIFs Correlates with Both Spontaneous and CK-666-Induced Cell Blebbing
3.3. Asymmetric Subcellular Distribution of VIFs Correlates with Biased Blebbing Activity at the Cell Edges
3.4. Initial Distribution of VIFs Predicts Blebbing Cell Behavior after CK-666 Application
3.5. Blebbing Activity in Mitotic Cells Correlates with Low Abundance of VIFs at the Cell Periphery
3.6. Enforced Removal of VIFs from the Cell Periphery Promotes Blebbing
3.7. Genetic Vimentin Depletion Stimulates Plasma Membrane Blebbing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Charras, G.T.; Yarrow, J.C.; Horton, M.A.; Mahadevan, L.; Mitchison, T.J. Non-equilibration of hydrostatic pressure in blebbing cells. Nature 2005, 435, 365–369. [Google Scholar] [CrossRef]
- Paluch, E.K.; Raz, E. The role and regulation of blebs in cell migration. Curr. Opin. Cell Biol. 2013, 25, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.T.; Hu, C.K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.; Paluch, E. Blebs lead the way: How to migrate without lamellipodia. Nat. Rev. Mol. Cell Biol. 2008, 9, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Sedzinski, J.; Biro, M.; Oswald, A.; Tinevez, J.Y.; Salbreux, G.; Paluch, E. Polar actomyosin contractility destabilizes the position of the cytokinetic furrow. Nature 2011, 476, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.C.; Stone, N.L.; Pittman, R.N. Extranuclear apoptosis. The role of the cytoplasm in the execution phase. J. Cell Biol. 1999, 146, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Trinkaus, J.P. Microvilli and blebs as sources of reserve surface membrane during cell spreading. Exp. Cell Res. 1976, 99, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Vargas, P.; Barbier, L.; Saez, P.J.; Piel, M. Mechanisms for fast cell migration in complex environments. Curr. Opin. Cell Biol. 2017, 48, 72–78. [Google Scholar] [CrossRef]
- Paluch, E.K.; Aspalter, I.M.; Sixt, M. Focal adhesion-independent cell migration. Annu. Rev. Cell Dev. Biol. 2016, 32, 469–490. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 2003, 3, 362–374. [Google Scholar] [CrossRef]
- Kosla, J.; Pankova, D.; Plachy, J.; Tolde, O.; Bicanova, K.; Dvorak, M.; Rosel, D.; Brabek, J. Metastasis of aggressive amoeboid sarcoma cells is dependent on Rho/ROCK/MLC signaling. Cell Commun. Signal 2013, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Giannoni, E.; Morandi, A.; Ippolito, L.; Ramazzotti, M.; Callari, M.; Gandellini, P.; Chiarugi, P. Mesenchymal to amoeboid transition is associated with stem-like features of melanoma cells. Cell Commun. Signal 2014, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Devreotes, P.N.; Zigmond, S.H. Chemotaxis in eukaryotic cells: A focus on leukocytes and Dictyostelium. Annu. Rev. Cell Biol. 1988, 4, 649–686. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Weigelin, B. Interstitial leukocyte migration and immune function. Nat. Immunol. 2008, 9, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Lammermann, T.; Sixt, M. Mechanical modes of ‘amoeboid’ cell migration. Curr. Opin. Cell Biol. 2009, 21, 636–644. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Tube travel: The role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008, 68, 7247–7249. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Petrie, R.J.; Yamada, K.M. Fibroblasts lead the way: A unified view of 3D cell motility. Trends Cell Biol. 2015, 25, 666–674. [Google Scholar] [CrossRef]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; Von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Brocker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar] [CrossRef]
- Bergert, M.; Chandradoss, S.D.; Desai, R.A.; Paluch, E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc. Natl. Acad. Sci. USA 2012, 109, 14434–14439. [Google Scholar] [CrossRef]
- Carragher, N.O.; Walker, S.M.; Scott Carragher, L.A.; Harris, F.; Sawyer, T.K.; Brunton, V.G.; Ozanne, B.W.; Frame, M.C. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: A link to integrin function. Oncogene 2006, 25, 5726–5740. [Google Scholar] [CrossRef] [PubMed]
- Ehrbar, M.; Sala, A.; Lienemann, P.; Ranga, A.; Mosiewicz, K.; Bittermann, A.; Rizzi, S.C.; Weber, F.E.; Lutolf, M.P. Elucidating the role of matrix stiffness in 3D cell migration and remodeling. Biophys. J. 2011, 100, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Le Berre, M.; Lautenschlaeger, F.; Maiuri, P.; Callan-Jones, A.; Heuze, M.; Takaki, T.; Voituriez, R.; Piel, M. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 2015, 160, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, E.; Poincloux, R.; Gauffre, F.; Maridonneau-Parini, I.; Le Cabec, V. Matrix architecture dictates three-dimensional migration modes of human macrophages: Differential involvement of proteases and podosome-like structures. J. Immunol. 2010, 184, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Beckham, Y.; Vasquez, R.J.; Stricker, J.; Sayegh, K.; Campillo, C.; Gardel, M.L. Arp2/3 inhibition induces amoeboid-like protrusions in MCF10A epithelial cells by reduced cytoskeletal-membrane coupling and focal adhesion assembly. PLoS ONE 2014, 9, e100943. [Google Scholar] [CrossRef]
- Chikina, A.S.; Svitkina, T.M.; Alexandrova, A.Y. Time-resolved ultrastructure of the cortical actin cytoskeleton in dynamic membrane blebs. J. Cell Biol. 2019, 218, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Chikina, A.S.; Rubtsova, S.N.; Lomakina, M.E.; Potashnikova, D.M.; Vorobjev, I.A.; Alexandrova, A.Y. Transition from mesenchymal to bleb-based motility is predominantly exhibited by CD133-positive subpopulation of fibrosarcoma cells. Biol. Cell 2019, 111, 245–261. [Google Scholar] [CrossRef]
- Derivery, E.; Fink, J.; Martin, D.; Houdusse, A.; Piel, M.; Stradal, T.E.; Louvard, D.; Gautreau, A. Free Brick1 is a trimeric precursor in the assembly of a functional wave complex. PLoS ONE 2008, 3, e2462. [Google Scholar] [CrossRef]
- Obeidy, P.; Ju, L.A.; Oehlers, S.H.; Zulkhernain, N.S.; Lee, Q.; Galeano Nino, J.L.; Kwan, R.Y.; Tikoo, S.; Cavanagh, L.L.; Mrass, P.; et al. Partial loss of actin nucleator actin-related protein 2/3 activity triggers blebbing in primary T lymphocytes. Immunol. Cell Biol. 2020, 98, 93–113. [Google Scholar] [CrossRef]
- Parri, M.; Taddei, M.L.; Bianchini, F.; Calorini, L.; Chiarugi, P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009, 69, 2072–2081. [Google Scholar] [CrossRef]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Balzer, E.M.; Tong, Z.; Paul, C.D.; Hung, W.C.; Stroka, K.M.; Boggs, A.E.; Martin, S.S.; Konstantopoulos, K. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 2012, 26, 4045–4056. [Google Scholar] [CrossRef] [PubMed]
- Holle, A.W.; Govindan Kutty Devi, N.; Clar, K.; Fan, A.; Saif, T.; Kemkemer, R.; Spatz, J.P. Cancer cells Invade confined microchannels via a self-directed mesenchymal-to-amoeboid transition. Nano Lett. 2019, 19, 2280–2290. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.D.; Mistriotis, P.; Konstantopoulos, K. Cancer cell motility: Lessons from migration in confined spaces. Nat. Rev. Cancer 2017, 17, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Niggemann, B.; Drell, T.L.T.; Joseph, J.; Weidt, C.; Lang, K.; Zaenker, K.S.; Entschladen, F. Tumor cell locomotion: Differential dynamics of spontaneous and induced migration in a 3D collagen matrix. Exp. Cell Res. 2004, 298, 178–187. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Cytoplasmic intermediate filaments in cell biology. Annu. Rev. Cell Dev. Biol. 2018, 34, 1–28. [Google Scholar] [CrossRef]
- Cheng, F.; Eriksson, J.E. Intermediate filaments and the regulation of cell motility during regeneration and wound healing. Cold Spring Harb. Perspect. Biol. 2017, 9, a022046. [Google Scholar] [CrossRef]
- Lowery, J.; Kuczmarski, E.R.; Herrmann, H.; Goldman, R.D. Intermediate filaments play a pivotal role in regulating cell architecture and function. J. Biol. Chem. 2015, 290, 17145–17153. [Google Scholar] [CrossRef]
- Sanghvi-Shah, R.; Weber, G.F. Intermediate filaments at the junction of mechanotransduction, migration, and development. Front. Cell Dev. Biol. 2017, 5, 81. [Google Scholar] [CrossRef]
- Serres, M.P.; Samwer, M.; Truong Quang, B.A.; Lavoie, G.; Perera, U.; Gorlich, D.; Charras, G.; Petronczki, M.; Roux, P.P.; Paluch, E.K. F-actin interactome reveals vimentin as a key regulator of actin organization and cell mechanics in mitosis. Dev. Cell 2020, 52, 210–222.e7. [Google Scholar] [CrossRef]
- Holwell, T.A.; Schweitzer, S.C.; Evans, R.M. Tetracycline regulated expression of vimentin in fibroblasts derived from vimentin null mice. J. Cell Sci. 1997, 110 Pt 16, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Colucci-Guyon, E.; Portier, M.M.; Dunia, I.; Paulin, D.; Pournin, S.; Babinet, C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell 1994, 79, 679–694. [Google Scholar] [CrossRef] [PubMed]
- Matveeva, E.A.; Venkova, L.S.; Chernoivanenko, I.S.; Minin, A.A. Vimentin is involved in regulation of mitochondrial motility and membrane potential by Rac1. Biol. Open 2015, 4, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Moir, R.D.; Prahlad, V.; Goldman, R.D. Motile properties of vimentin intermediate filament networks in living cells. J. Cell Biol. 1998, 143, 147–157. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Helfand, B.T.; Mendez, M.G.; Murthy, S.N.; Shumaker, D.K.; Grin, B.; Mahammad, S.; Aebi, U.; Wedig, T.; Wu, Y.I.; Hahn, K.M.; et al. Vimentin organization modulates the formation of lamellipodia. Mol. Biol. Cell 2011, 22, 1274–1289. [Google Scholar] [CrossRef] [PubMed]
- Chernoivanenko, I.S.; Matveeva, E.A.; Gelfand, V.I.; Goldman, R.D.; Minin, A.A. Mitochondrial membrane potential is regulated by vimentin intermediate filaments. FASEB J. 2015, 29, 820–827. [Google Scholar] [CrossRef]
- Svitkina, T. Imaging cytoskeleton components by electron microscopy. Methods Mol. Biol. 2016, 1365, 99–118. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Rovensky, Y.A.; Bershadsky, A.D.; Vasiliev, J.M. Transverse pattern of microfilament bundles induced in epitheliocytes by cylindrical substrata. J. Cell Sci. 1995, 108, 735–745. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Borisy, G.G. Correlative light and electron microscopy of the cytoskeleton of cultured cells. Methods Enzymol. 1998, 298, 570–592. [Google Scholar] [CrossRef]
- Svitkina, T.M. Platinum replica electron microscopy: Imaging the cytoskeleton globally and locally. Int. J. Biochem. Cell Biol. 2017, 86, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T.M.; Verkhovsky, A.B.; Borisy, G.G. Improved procedures for electron microscopic visualization of the cytoskeleton of cultured cells. J. Struct. Biol. 1995, 115, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D. The role of three cytoplasmic fibers in BHK-21 cell motility. I. Microtubules and the effects of colchicine. J. Cell Biol. 1971, 51, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Grin, B.; Mahammad, S.; Wedig, T.; Cleland, M.M.; Tsai, L.; Herrmann, H.; Goldman, R.D. Withaferin a alters intermediate filament organization, cell shape and behavior. PLoS ONE 2012, 7, e39065. [Google Scholar] [CrossRef] [PubMed]
- Messica, Y.; Laser-Azogui, A.; Volberg, T.; Elisha, Y.; Lysakovskaia, K.; Eils, R.; Gladilin, E.; Geiger, B.; Beck, R. The role of vimentin in regulating cell invasive migration in dense cultures of breast carcinoma cells. Nano Lett. 2017, 17, 6941–6948. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, D.; Jones, J.C. The vimentin cytoskeleton regulates focal contact size and adhesion of endothelial cells subjected to shear stress. J. Cell Sci. 2003, 116, 4977–4984. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.T.; Coughlin, M.; Mitchison, T.J.; Mahadevan, L. Life and times of a cellular bleb. Biophys. J. 2008, 94, 1836–1853. [Google Scholar] [CrossRef]
- Lavenus, S.B.; Tudor, S.M.; Ullo, M.F.; Vosatka, K.W.; Logue, J.S. A flexible network of vimentin intermediate filaments promotes migration of amoeboid cancer cells through confined environments. J. Biol. Chem. 2020, 295, 6700–6709. [Google Scholar] [CrossRef]
- Patteson, A.E.; Pogoda, K.; Byfield, F.J.; Mandal, K.; Ostrowska-Podhorodecka, Z.; Charrier, E.E.; Galie, P.A.; Deptula, P.; Bucki, R.; McCulloch, C.A.; et al. Loss of vimentin enhances cell motility through small confining spaces. Small 2019, 15, e1903180. [Google Scholar] [CrossRef]
- Tyson, R.A.; Zatulovskiy, E.; Kay, R.R.; Bretschneider, T. How blebs and pseudopods cooperate during chemotaxis. Proc. Natl. Acad. Sci. USA 2014, 111, 11703–11708. [Google Scholar] [CrossRef]
- Rentsch, P.S.; Keller, H. Suction pressure can induce uncoupling of the plasma membrane from cortical actin. Eur. J. Cell Biol. 2000, 79, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.Y.; Schulze, U.; Salbreux, G.; Roensch, J.; Joanny, J.F.; Paluch, E. Role of cortical tension in bleb growth. Proc. Natl. Acad. Sci. USA 2009, 106, 18581–18586. [Google Scholar] [CrossRef] [PubMed]
- Esue, O.; Carson, A.A.; Tseng, Y.; Wirtz, D. A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem. 2006, 281, 30393–30399. [Google Scholar] [CrossRef] [PubMed]
- Svitkina, T.M.; Verkhovsky, A.B.; Borisy, G.G. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 1996, 135, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Lanier, M.H.; Kim, T.; Cooper, J.A. CARMIL2 is a novel molecular connection between vimentin and actin essential for cell migration and invadopodia formation. Mol. Biol. Cell 2015, 26, 4577–4588. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Viedma-Poyatos, A.; Navarro-Carrasco, E.; Martinez, A.E.; Pajares, M.A.; Perez-Sala, D. Vimentin filaments interact with the actin cortex in mitosis allowing normal cell division. Nat. Commun. 2019, 10, 4200. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Shen, Y.; Sivagurunathan, S.; Weber, M.S.; Adam, S.A.; Shin, J.H.; Fredberg, J.J.; Medalia, O.; Goldman, R.; Weitz, D.A. Vimentin intermediate filaments and filamentous actin form unexpected interpenetrating networks that redefine the cell cortex. Proc. Natl. Acad. Sci. USA 2022, 119, e2115217119. [Google Scholar] [CrossRef]
- Mendez, M.G.; Restle, D.; Janmey, P.A. Vimentin enhances cell elastic behavior and protects against compressive stress. Biophys. J. 2014, 107, 314–323. [Google Scholar] [CrossRef]
- Wang, N.; Stamenovic, D. Contribution of intermediate filaments to cell stiffness, stiffening, and growth. Am. J. Physiol. Cell Physiol. 2000, 279, C188–C194. [Google Scholar] [CrossRef]
- Jiu, Y.; Peranen, J.; Schaible, N.; Cheng, F.; Eriksson, J.E.; Krishnan, R.; Lappalainen, P. Vimentin intermediate filaments control actin stress fiber assembly through GEF-H1 and RhoA. J. Cell Sci. 2017, 130, 892–902. [Google Scholar] [CrossRef]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, N.B.M.; Nishimura, Y.; Plotnikov, S.V.; Thiagarajan, V.; Zhang, Z.; Shi, S.; Natarajan, M.; Viasnoff, V.; Kanchanawong, P.; Jones, G.E.; et al. A mechano-signalling network linking microtubules, myosin IIA filaments and integrin-based adhesions. Nat. Mater. 2019, 18, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Maeda, F.; Nagasako, T.; Mochizuki, Y.; Uchida, S.; Ikenouchi, J. A RhoA and Rnd3 cycle regulates actin reassembly during membrane blebbing. Proc. Natl. Acad. Sci. USA 2016, 113, E1863–E1871. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Wu, D.; Wei, Q. Myosin II-interacting guanine nucleotide exchange factor promotes bleb retraction via stimulating cortex reassembly at the bleb membrane. Mol. Biol. Cell 2018, 29, 643–656. [Google Scholar] [CrossRef]
- Guo, M.; Ehrlicher, A.J.; Mahammad, S.; Fabich, H.; Jensen, M.H.; Moore, J.R.; Fredberg, J.J.; Goldman, R.D.; Weitz, D.A. The role of vimentin intermediate filaments in cortical and cytoplasmic mechanics. Biophys. J. 2013, 105, 1562–1568. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chikina, A.S.; Zholudeva, A.O.; Lomakina, M.E.; Kireev, I.I.; Dayal, A.A.; Minin, A.A.; Maurin, M.; Svitkina, T.M.; Alexandrova, A.Y. Plasma Membrane Blebbing Is Controlled by Subcellular Distribution of Vimentin Intermediate Filaments. Cells 2024, 13, 105. https://0-doi-org.brum.beds.ac.uk/10.3390/cells13010105
Chikina AS, Zholudeva AO, Lomakina ME, Kireev II, Dayal AA, Minin AA, Maurin M, Svitkina TM, Alexandrova AY. Plasma Membrane Blebbing Is Controlled by Subcellular Distribution of Vimentin Intermediate Filaments. Cells. 2024; 13(1):105. https://0-doi-org.brum.beds.ac.uk/10.3390/cells13010105
Chicago/Turabian StyleChikina, Aleksandra S., Anna O. Zholudeva, Maria E. Lomakina, Igor I. Kireev, Alexander A. Dayal, Alexander A. Minin, Mathieu Maurin, Tatyana M. Svitkina, and Antonina Y. Alexandrova. 2024. "Plasma Membrane Blebbing Is Controlled by Subcellular Distribution of Vimentin Intermediate Filaments" Cells 13, no. 1: 105. https://0-doi-org.brum.beds.ac.uk/10.3390/cells13010105