
The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases


1
Departamento de Bioquímica y Biología Molecular y Genética, Facultad de Enfermería y Terapia Ocupacional, Universidad de Extremadura, 10003 Cáceres, Spain
2
Centro de Investigación Biomédica en Red en Enfermedades Neurodegenerativas, Instituto de Salud Carlos III (CIBER-CIBERNED-ISCIII), 28029 Madrid, Spain
3
Instituto Universitario de Investigación Biosanitaria de Extremadura (INUBE), 10003 Cáceres, Spain
4
Departmentof Neurology, Columbia University, New York, NY 10032, USA
*
Authors to whom correspondence should be addressed.
Cells 2024, 13(7), 609; https://0-doi-org.brum.beds.ac.uk/10.3390/cells13070609
Submission received: 18 February 2024
/
Revised: 28 March 2024
/
Accepted: 29 March 2024
/
Published: 30 March 2024
(This article belongs to the Special Issue Mitochondria in Developmental and Age-Associated Neurodegenerative Diseases)
Abstract
:Cardiolipin (CL) is a mitochondria-exclusive phospholipid synthesized in the inner mitochondrial membrane. CL plays a key role in mitochondrial membranes, impacting a plethora of functions this organelle performs. Consequently, it is conceivable that abnormalities in the CL content, composition, and level of oxidation may negatively impact mitochondrial function and dynamics, with important implications in a variety of diseases. This review concentrates on papers published in recent years, combined with basic and underexplored research in CL. We capture new findings on its biological functions in the mitochondria, as well as its association with neurodegenerative diseases such as Alzheimer’s disease or Parkinson’s disease. Lastly, we explore the potential applications of CL as a biomarker and pharmacological target to mitigate mitochondrial dysfunction.
1. Introduction
Cardiolipin (CL; 1,3-bis(sn-3′-phosphatidyl)-sn-glycerol) was isolated from beef hearts by Mary C. Pangborn in the 1940s [1]. It is predominantly localized and synthesized in the inner mitochondrial membrane (IMM) and constitutes approximately 15–20% of the total mitochondrial phospholipids [2]. It has a conical shape characterized by a double glycerophosphate backbone, three glycerol moieties, and four fatty acyl (FA) chains (rather than two fatty acyl side chains typically observed in phospholipid structures) which can show different lengths or degrees of saturation. It exerts lateral pressure within the membrane causing negative membrane curvature [3]. De novo CL biosynthesis in mammals is a multi-step process that begins with the synthesis and transport of phosphatidic acid (PA) from the endoplasmic reticulum (ER) to mitochondria. Alternatively, mitochondrial phospholipase D (MitoPLD) can hydrolyze CL to generate PA in the OMM [4]. Next, PA is translocated to the IMM where several enzymes are involved in CL biosynthesis, including TAM41 mitochondrial translocator assembly and maintenance homolog (TAMM41); phosphatidylglycerol phosphate synthase (PGS1); protein-tyrosine phosphatase mitochondrial 1 (PTPMT1); and CL synthase (CLS1), which catalyzes the formation of nascent CL (CLn). CLn, which is characterized by the presence of saturated acyl chains, is remodeled by phospholipase A2 (PLA2) into the transient intermediate phospholipid monolysocardiolipin (MLCL), which is subsequently re-acylated by TAFAZZIN (tafazzin phospholipid remodeling enzyme) to form mature CL (CLm), which is enriched in unsaturated FA chains. Furthermore, CL remodeling can also occur via the acyl transferases ALCAT1 or MLCLAT1 [5]. However, the extent to which these alternative pathways contribute to CL remodeling is still under debate. A schematic representation of the synthesis and remodeling of CL is depicted in Figure 1.
CL exhibits the greatest structural diversity among all phospholipid classes due to its tetra-acylated structure. Notably, the arrangement of fatty acyl chains in CL is not stochastic. Each CL contains unique lengths or degrees of unsaturation in its fatty acid (FA) tails through post-synthetic remodeling, which appears to be tissue- and cell-specific. For instance, in the heart, the composition of CL is restricted to one dominant FA, the linoleic acid (18:2), resulting in a homogeneous population, whereas, in the brain, oleic acid (18:1) along with longer-chain unsaturated FA such as arachidonic acid (20:4) and docosahexaenoic acid (22:6) are the predominant side chains [6,7].
Neurodegenerative diseases affect millions of people globally, and, currently, treatments are very limited. While many of these diseases involve the misfolding and aggregation of proteins, leading to the formation of insoluble fibrils, tangles, and plaques, these findings alone almost certainly cannot explain the clinical diversity observed in these disorders. Intriguingly, mitochondrial dysfunction is strongly implicated in the pathogenesis of numerous neurodegenerative diseases such as Alzheimer’s disease (AD) [8], or Parkinson’s disease (PD) [9]. However, in most cases, how aberrant mitochondrial function leads to disease and phenotypes is unclear. Remarkably, recent studies have shown that alterations in mitochondrial homeostasis associated with neurodegeneration are influenced by mitochondrial lipid composition and overall lipid metabolism [10]. For this reason, there is a growing interest in the use of lipids as potential biomarkers for neurodegenerative diseases. Specifically, quantitative and structural changes in CL have been shown in the context of neurodegenerative diseases [11,12,13]. The identification of CL as a candidate biomarker in diagnosing neurodegenerative diseases has been supported by recent advancements in lipidomics studies using human blood from patients and animal models [14,15,16]. While the role of CL in mitochondria has been discussed in other reviews [17,18], here, we focus on new discoveries that appeared in recent years, coupled with basic and underexplored research in CL. Specifically, we explore new biological functions and the proteins affected by changes in CL content, leading to mitochondrial dysfunction. We discuss the role of CL in the pathology of neurodegenerative disorders and its use as a biomarker. Finally, we address the discovery of recent CL-binders and their potential role as therapeutic agents to ameliorate mitochondrial dysfunction and prevent neurodegeneration.
2. Physicochemical Properties and Detection of CL
The structure of CL differs from other phospholipids because two phosphatidates bind to a single central glycerol headgroup, resulting in a conical shape with a smaller cross-sectional area in the headgroup which restricts its mobility to interact with other phospholipid head groups. As a cone shape, with the polar region at the top and the flexible acyl chains at the base of the “cone”, it aggregates in structures with a negative curvature in the IMM facing the cristae structures. On the contrary, the positively curved outer monolayer leaflet primarily consists of phosphatidylcholine. Besides its preference for forming a non-bilayer structure, such as the hexagonal phase, CL can also adopt a bilayer structure with a preference for the lamellar phase in the presence of divalent cations [19,20]. In addition, the distribution of CL is determined by the lipid-scaffolding proteins prohibitin-1 and prohibitin-2, which may facilitate the remodeling process as part of the biosynthetic machinery [21].
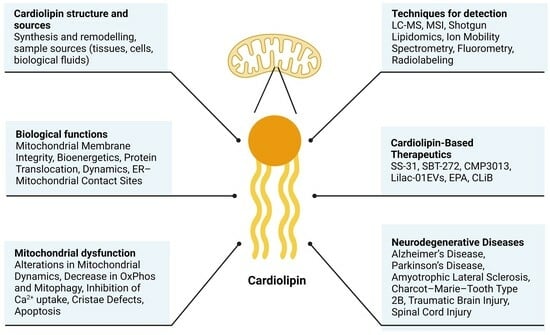
The detection and quantification of CL constitute valuable tools for verifying mitochondrial dysfunction, characterizing the pathophysiological mechanisms of disease, and facilitating clinical diagnostics. Several methods are commonly used to analyze CL, mainly liquid chromatography combined with mass spectrometry (LC-MS), mass spectrometry imaging (MSI), shotgun lipidomics, ion mobility spectrometry (IMS), fluorometry, and radiolabeling (Figure 2). Before the analysis, it is necessary to extract and isolate the lipid content, unless fluorescent dyes such as 10-N-Nonyl acridine orange (NAO) are utilized, which can directly stain cells or tissues [22]. Mass spectrometric techniques are increasingly accessible and offer numerous advantages, particularly in sensitivity and specificity, over older chromatographic and fluorometric methods. In recent years, LC-MS-based multi-analyte/lipidomic assays have also been utilized in clinical practice.
Different biological samples can be employed in investigating the role of CL in health and disease. Samples include tissues such as brain, liver, or heart, and cell lines such as patient-derived fibroblasts, as well as biological fluids such as serum, urine, or cerebrospinal fluid. Moreover, induced pluripotent stem cells differentiated into disease-relevant cell types, including cortical neurons, myotubes, or cardiomyocytes, enable researchers to characterize CL in tissue-specific human disease models [23,24]. The specific samples studied in each disease are detailed in Section 5. Given the unavailability of brain biopsy samples from living patients, samples collected from blood, urine, or saliva can be used as an alternative to identify “CL signatures” capable of predicting the severity of the disease.
3. The Role of Cardiolipin in Mitochondrial Biological Functions
The structure features of CL allow it to play a crucial role in various mitochondrial processes, including the membrane architecture, the stabilization of the mitochondrial respiratory chain, the assembly of F1-F0 ATP synthase, the organization of supercomplexes, the regulation of mitochondrial dynamics, and ER–mitochondrial contact sites. Next, we describe new findings about the role of CL in mitochondrial biological functions.
3.1. The Role of Cardiolipin in Mitochondrial Membrane Integrity
The structural and dynamic characteristics of mitochondrial membranes are significantly influenced by the biophysical properties of CL. An elevated CL concentration increases the lateral diffusion of lipids, affecting the mitochondria membrane composition [25]. The increased dipole potential observed in the IMM can be attributed to its elevated CL content in contrast to the outer mitochondrial membrane (OMM) [26]. Thus, CL may play a crucial role in controlling the flux rate of protons and ions during ATP production. Additionally, CL, characterized by a cone-like shape, accumulates in curved regions of the membrane, with higher sorting to more highly curved and negative membranes [3]. Apart from the biophysical effect of CL on mitochondrial membranes, CL is crucial for the formation and maintenance of the mitochondrial tubular cristae. Although the molecular mechanisms shaping cristae are becoming understood, the synergic contribution of three major protein complexes, OPA1, F1-F0 ATP synthase, and the MICOS complex, are recognized as essential for the formation and maintenance of cristae in eukaryotic cells [27]. It is well established that the cristae-forming protein complexes, F1-F0 ATP synthase, and the MICOS complex are stabilized by CL, which promotes cristae formation [28,29]. Specifically, CL binds to the subunit MIC27, which is part of the MIC10 subcomplex, preserving the integrity of the MICOS complex at the cristae [29,30,31,32]. Moreover, CL regulates the ATP synthase activity [33,34] (see next section for more details).
3.2. Role of Cardiolipin in Bioenergetics
CL is thought to act as a glue holding the respiratory supercomplex (RSC), high-molecular-weight structures consisting of complexes I, III, and IV in mammals [35]. RSCs are proposed to assist in the electron transfer between individual respiratory chain components acting to lessen oxidative damage [36]. Thus, CL plays an important role in the integrity of the RSC, reducing ROS generation [24]. CL also plays a role in anchoring cytochrome c to the IMM and facilitates the electron transfer from complex III to complex IV [37]. In addition, CL supports efficient OXPHOS activity. Recent cryogenic electron microscopy studies in bovine mitochondria have also revealed that CL binds tightly to F1-F0 ATP synthase and stabilizes its interaction with the IMM, thus controlling proton leakage and improving ATP generation, in addition to participating in dimer assembly [34]. Additionally, theoretical simulations show that the transient binding of CL to F1-F0 ATP synthase lubricates its rotor [33]. Finally, a recent study has shown that the mitochondrial ribosome binds CL, thereby stabilizing the IMM association of the protein translation machinery and supporting the biogenesis of mitochondrial OXPHOS proteins [38].
3.3. Role of Cardiolipin in Mitochondrial Protein Translocation
CL is also indispensable for the stability, assembly, and activity of mitochondrial carrier proteins and ion channels. Specifically, it plays a crucial role in shaping the conformation and assembly of the ADP/ATP carrier protein (AAC) which resides in the IMM and exchanges ADP and ATP to enable OXPHOS [39,40]. CL is also vital for the assembly and functioning of the mitochondrial Ca2+ uniporter (MCU) complex, which co-ordinates cellular energy demands with mitochondrial bioenergetics through Ca2+ signaling [41]. CryoEM structures of the MCU holo-complex demonstrate that MCU tetramers are stabilized by eight CL molecules [42,43]. CL is also necessary for activating various mitochondrial proteins, such as pyruvate dehydrogenase [44], and frataxin, which are crucial for the biogenesis of mitochondrial iron–sulfur clusters [45].
3.4. Role of Cardiolipin in Mitochondrial Dynamics
Mitochondria are dynamic organelles orchestrating continuous cycles of fusion and division, crucial for regulating mitochondrial size, number, distribution, function, and turnover [46]. CL plays a role in the maintenance of mitochondrial dynamics by regulating the fusion and fission machinery. It is known that CL interacts with DRP1 (dynamin-related protein 1) promoting mitochondrial constriction [47]. Conversely, it has been shown that the externalization of CL in the outer leaflet of the IMM plays an important role in Opa1-mediated efficiency fusion [48]. Human OPA1 is embedded into CL-containing membranes through a lipid-binding paddle domain [49].
3.5. Role of Cardiolipin in ER–Mitochondrial Contact Sites
The communication between the ER and mitochondria takes place through their networks, both physically and functionally [50]. These interactions, referred to as mitochondria-associated ER membranes (MAMs), play a crucial role in transporting lipids from the ER, the primary site of lipid biosynthesis, to the mitochondria [51]. New findings indicate that CL plays a role in regulating ER–mitochondrial contact sites. First, a recent study has shown that the tyrosine phosphatase-interacting protein 5 (1PTPIP51) binds and transfers PA from the ER to mitochondria at MAM, and its deletion resulted in reduced mitochondrial CL levels [52]. PTPIP51 also interacts with VAPB (vesicle-associated membrane protein-associated protein B), an ER protein, to form a tether between the ER and the mitochondria [52]. Second, both the enzyme ALCAT1 and the lipid CL have been found to be enriched at MAM [53]. Third, MICOS subcomplexes assemble in proximity to ER contact sites [54]. Together, these studies provide evidence that CL metabolism may be modulated at ER–mitochondrial contact sites, thereby influencing their function. Consequently, disruptions in ER–mitochondrial connectivity may directly contribute to mitochondrial ultrastructural disorganization, leading to disorders associated with CL homeostasis.
3.6. Other Functions
Recently, the discovery of new interactions between CL and other proteins suggests that CL is involved in additional activities. For instance, CL showed extremely higher affinity for the potassium channel KcsA (K+ channel, Streptomyces lividans A) than for monoanionic lipids [55]. Furthermore, CL regulates the activity of the mitochondrial ABC transporter 10 (ABCB10), which is an exporter localized in the IMM [56].
4. The Role of Cardiolipin Alterations in Mitochondrial Dysfunction
Pathological alterations in CL profiles have been reported in several diseases, including loss of content, aberrant remodeling, and/or peroxidation. The reduction in CL content may result from increased CL degradation catalyzed by calcium-independent phospholipase A2γ (iPLA2γ) [57]. Alterations/mutations in enzymes responsible for CL biosynthesis or remodeling lead to aberrant CL [58]. For instance, mutations in TAFAZZIN lead to CL deficiency in the cardiomyopathy Barth syndrome [59]. Additionally, CL is highly susceptible to oxidation due to its elevated content of unsaturated fatty acids, mainly linoleic acid, and its proximity to the electron transport chain (ETC), the site of reactive oxygen species (ROS) production [60]. Under physiological conditions, approximately 15–20% of cytochrome c binds to CL in the IMM [61]. CL can translocate to the OMM when ROS are present, thereby effectively increasing the likelihood of CL binding to cytochrome c. Subsequently, cytochrome c, which normally acts as a shuttle of electrons between respiratory complexes III and IV, becomes a CL peroxidase, capable of catalyzing CL oxidation and peroxidation reactions [62]. In this process, it is important to mention that the remodeling of cristae structures at the IMM has been demonstrated to facilitate the release of cytochrome c [63]. Interestingly, a recent study has found that cytochrome c can induce CL peroxidation independent of ROS, which is controlled by its redox states [63]. The authors hypothesize that a two-step process (the ROS-independent and ROS-dependent steps) might be involved in the early stage of apoptosis before cytochrome c release [63], although further studies are required to demonstrate this statement.
The oxidation of CL leads to conformational changes that affect the physical properties of the IMM and OXPHOS activity [64]. Oxidized CL acts as a signaling molecule to promote mitophagy (mitochondrial degradation) and/or apoptosis (programmed cell death). During mitophagy, CL traverses the IMM to arrive at the OMM in a process facilitated by the mitochondrial nucleoside diphosphokinase [65] and/or phospholipid scramblase 3 [66]. Once externalized, CL recruits microtubule-associated protein 1A/1B-light chain (LC3) [66]. The N-terminal variability of the LC3 protein subfamily determines its specificity for CL. Thus, the LC3A (MAP1LC3A) exhibits a high affinity and specificity for CL, actively participating in CL-mediated mitophagy [67]. LC3C (MAP1LC3C) has the highest affinity for CL but does not partake in CL-mediated mitophagy. Instead, it is proposed that LC3C’s binding to PA on the OMM contributes to maintaining the integrity of the mitochondrial network under normal growth conditions through the selective degradation of mitochondrial proteins [67]. During apoptosis, the externalization of oxidized CL promotes the increase in caspase-3 activity and Bcl-2–associated X protein (Bax) [67,68]. Therefore, alterations in the CL profile result in the malfunction of proteins and enzymes, leading to mitochondrial dysfunction and disease. Relevant proteins affected by CL dysfunction leading to mitochondrial alterations are MIC10 subunit [29,30,31], F1Fo ATP synthase [28,69], RCs [70], ADP/ATP carrier [39], MCU [41,42], PARL [71], DRP1 [47] [72], OPA1 [48], LC3 [67] and cytochrome c [73]. A full list of proteins affected by CL dysfunction, alongwith their corresponding mitochondrial alterations in each biological model, is summarized in Table 1 and represented in Figure 3.
5. The Role of Cardiolipin in Neurodegenerative Diseases
The brain exhibits a CL profile with the highest molecular diversity, comprising up to 100 different species [6,74,75]. This extraordinary diversification may be attributed to its various functions. It is believed that the role of CL in structural organization is facilitated by the presence of non-PUFA species (with saturated and monounsaturated acyls) and symmetric linoleoyl-containing CL species, thus leading to its predominant presence in mammalian tissues with high bioenergetic demands, such as the heart and skeletal muscles. Conversely, the role of CL in the signaling function is thought to be carried out by longer-chain PUFA-containing species, as found in the brain [74,76,77]. At the cellular level, it has been found that the content of CL is roughly two-fold higher in glial cells compared to neurons [78]. This difference in CL levels may contribute to variations in intracellular signaling molecules and functional activity between these two primary types of brain cells [78]. Interestingly, CL content is reduced in non-synaptic mitochondria compared to synaptic mitochondria from the brains of aged rats [79]. The distinct mechanisms by which the tissue- and cell-specificity of CL compositions are generated and regulated are still largely elusive.
Given that the brain consumes over 20% of the total body energy, CL abnormalities, including changes in the CL content, FA acyl chain composition, and CL oxidation in the CNS, are unsurprisingly linked to various neurodegenerative diseases. Consequently, gaining a comprehensive understanding of the role of defective CL metabolism in CNS homeostasis and brain function is likely to provide significant insights into the pathophysiology of neurodegenerative processes. The recent findings in CL-mediated neurodegenerative disorders are summarized as follows (see Table 2).
5.1. Alzheimer’s Disease (AD)
AD is the most prevalent adult neurodegenerative disorder. Pathologically, it manifests through progressive neuronal loss in the hippocampus and cortex, accompanied by the accumulation of extracellular neuritic plaques and intracellular neurofibrillary tangles in the brain. A prominent component within these plaques is β-amyloid (Aβ), formed through the cleavage of the amyloid precursor protein (APP) by presenilin-1 (PS1) and/or presenilin-2 (PS2), both integral to the γ-secretase complex [94]. Significantly, dominantly inherited mutations in both presenilins and APP currently represent the sole known causes of familial Alzheimer’s disease (FAD). This observation has led to the widely accepted “amyloid cascade” hypothesis, positing that the deposition of Aβ in the brain serves as the initiating pathological event in AD [95]. While this hypothesis elucidates the development of plaques and, potentially, tangles, it fails to explain other facets of the disease [96]. Recent studies propose an alternative perspective, suggesting that AD may be rooted in a disorder of lipid metabolism [97,98,99]. A low CL content associated with impaired mitochondrial synaptic function and oxidative stress has been reported in vivo [11], and in vitro [80]. In addition, a significant reduction of CL that contained polyunsaturated FA (PUFA) was observed in the cortex of human brains affected by AD [81]. Both in vivo and in vitro models of AD have demonstrated the high susceptibility of mitochondrial membranes to tau aggregates, resulting in the overexpression of tau aggregates, resulting in neuronal toxicity [100]. Tau protein exhibits a preference for binding to CL-rich regions of the OMM, triggering mitochondrial swelling, cytochrome c release, and a reduction in membrane potential in vitro [82]. Another aspect of AD is the dysregulated neuroinflammatory response, caused by persistent microglial activation and the release of proinflammatory cytokines. A potential protective role of CL in mitigating the AD inflammatory response has been postulated [83]. Finally, it has been demonstrated that alterations in the ATP-binding cassette (ABC) subfamily A member 7 gene (ABCA7), identified as a strong risk factor for late-onset AD, induce a reduction in CL content [84]. While recent advances in lipidomics have proposed other lipids as potential biomarkers in AD using blood samples from living patients and brain tissue from autopsy patients (reviewed by [101,102]), there have been no clinical studies evaluating the levels of CL in AD patients to date.
5.2. Parkinson’s Disease (PD)
PD is the most prevalent movement disorder and the second most common neurodegenerative disease after AD. Pathologically, PD is caused by the loss of dopaminergic neurons in the substantia nigra, a basal ganglia structure in the midbrain, which affects both cognitive function and gait. It is one of the synucleinopathies diseases, whose hallmark is represented by Lewy bodies, which are characterized by alpha-synuclein (α-syn) inclusions [103]. The protein α-syn is intrinsically disordered and it is highly expressed in neurons [104]. The majority of PD is idiopathic, with familial PD constituting 5–10% of cases. Recent data suggest that α-syn interacts with CL, potentially impacting the mitochondrial membrane integrity [85,105]. However, the pathomechanism behind this interaction is still unclear. OMM-localized CL promotes the refolding of disordered α-syn monomers to an α-helical structure, preventing neuronal loss and leading to mitophagy [23]. Moreover, α-syn can form a triple complex with CL and cytochrome c, which prevents cytochrome c release and delays neuronal damage [106]. This observation is in contrast to reports that anionic membranes promote α-syn aggregation [107]. Indirect evidence also suggests that α-syn oligomers interact with CL to favor pore formation within mitochondrial membranes affecting membrane permeability [86]. The formation of α-syn fibrils increased in the presence of CL leading to the unfolded protein response (UPR) in the ER and mitochondria [87]. Interestingly, the localization of α- syn at ER–mitochondrial contact sites was also reported in different models [108,109]. These studies have shown that the overexpression of α-syn disrupted the physical contacts and Ca2+ transfer between the two organelles, leading to alterations in mitochondrial metabolism. It is possible that aggregates of α-syn could, thus, directly disrupt mitochondrial metabolism via interactions with CL-rich microdomains or contact sites between the mitochondria and ER. The inhibition of the enzyme ALCAT1 may reduce α-syn oligomerization, preventing apoptosis [88]. On the other hand, no changes in the total CL content have been observed across various PD murine models [12] or PD patients [89]. A lipidomic analysis in rat rotenone models of PD revealed a notable loss of linoleic-acid-containing CL species and the accumulation of CL oxidation in the substantia nigra during later stages of exposure. In contrast, elevated levels of PUFA-containing CLs were detected in plasma, suggesting that non-apoptotic cell death pathways are also triggered in rotenone-treated animals [16]. These collective findings underscore the significant role of CL in PD, raising the possibility that CL species might be important biomarkers in neuropathological stages [101]. Whereas new lipidomic analysis using whole blood and serum from PD patients reveals that the progression of the disease could be predicted by a lipid-biomarker panel [110,111,112], CL is not yet included in those studies, similar to what we found in AD.
5.3. Amyotrophic Lateral Sclerosis (ALS)
ALS is a fatal neurological disorder characterized by the selective loss of motor neurons, resulting in muscle atrophy, paralysis, and respiratory failure. The exact cause of ALS remains uncertain, with around 90% of cases having unknown origins (termed as sporadic ALS), and the remaining 10% identified as hereditary cases (known as familial ALS). For familial ALS, 42 genes including SOD1 (superoxide dismutase 1), C9ORF72 (chromosome 9 open reading frame 72), TARDBP (tAR DNA-binding protein), and FUS (fus RNA-binding protein) have been identified to be causal factors [113]. Interestingly, there is growing evidence linking alterations in lipid metabolism to ALS pathogenesis [114], although its implications remain unknown. The mouse model of ALS with FUS overexpression exhibits high levels of cholesterol esters, specific ceramides, and the dysregulation of phospholipids, including reduced levels of cardiolipin [13]. Along this line, the reduction of CL levels in the spinal cord has also been observed in the transgenic rat model SOD1-G86R (superoxide dismutase 1 Gly86Arg), along with other lipidomic changes [14]. Notably, the spinal cord and, to a lesser extent, brain mitochondria of SOD1-G93A transgenic mice exhibit increased CL peroxidation, impaired OXPHOS activity, and heightened cytochrome c release from the IMM, aligning with the low binding affinity of oxidized CL for cytochrome c [115]. A recent study has shown no significant changes in CL in the serum from ALS patients compared to controls, but the authors observed an increase in the unsaturated FA, which may affect the different species of CL [90]. These emerging lines of evidence suggest that aberrant CL metabolism plays a broader role in ALS pathogenesis. Future studies will help to understand better the association between CL and ALS.
5.4. Charcot–Marie–Tooth Type 2B (CMT2B)
CMT2B is a rare hereditary peripheral neuropathy resulting from five missense mutations in the RAB7A (ras-related protein Rab-7a) gene, responsible for encoding a small GTPase within the RAB (ras-related in the brain) family. [116]. Currently, no cure is available for this disease. Pathologically, the CMT2B peripheral neuropathy is characterized by prominent sensory loss, progressive distal weakness leading to atrophy, reduced tendon reflexes, and normal or near-normal nerve conduction [116]. Notably, a recent lipidomic analysis has revealed changes in the lipidomic profile of CMT2B-derived fibroblasts versus healthy donor cells. The authors found that CMT2B cells exhibited an elevated level of monounsaturated FA and increased expression of key enzymes involved in monounsaturated and polyunsaturated FA synthesis [91]. Although the authors did not include the analysis of CL, it is conceivable that its content is also altered.
5.5. Traumatic Brain Injury
Traumatic brain injury (TBI) results from a sudden, external force causing damage to the brain. The severity of TBI can range from acute to chronic, leading to cognitive, emotional, and physical impairments. In fact, TBI is a leading cause of neurodegenerative diseases. Recent studies using mass spectrometric imaging have revealed that early phases of TBI exhibited reductions in CL levels in the contusional cortex, ipsilateral hippocampus, and thalamus, with the most highly unsaturated CL species being particularly vulnerable to loss [77]. Moreover, TBI is associated with an increase in mitophagy leading to the externalization of CL to the OMM, thereby eliminating mitochondrial damage and preventing neuronal death [92]. Interestingly, recent studies are investigating the detection of CL in animal and human biofluids like blood, urine, saliva, and tears for its potential role as a biomarker in diagnosing traumatic brain injury (TBI) [15,117].
5.6. Spinal Cord Injury
Traumatic spinal cord injury (SCI) results in neurological deficits below the level of injury. Currently, there is no effective treatment available for SCI patients. A recent research study has revealed a significant 50% decrease in CL species identified in adult rat spinal cord following a moderate contusive SCI [93]. The decreased CL species predominantly contained polyunsaturated fatty acids, making them highly susceptible to peroxidation. The authors also found that mitochondrial oxidative stress induced CL oxidation and led to CL loss by activating iPLA2γ to hydrolyze CL. These CL alterations induced mitochondrial dysfunction and subsequent neuronal death. Remarkably, simultaneous measurements of cytochrome c release, apoptotic protein Smac/DIABLO (second mitochondria-derived activator of caspases/direct IAP binding protein with low pI) release, and caspase-3 activity were used in this study as biomarkers to assess the extent of cardiolipin loss in rat models of SCI [93].
6. Cardiolipin-Based Therapeutics
The identification and characterization of novel CL-binder candidates that have the potential to attenuate mitochondrial damage is a priority. SS-31 (Szeto-Schiller-31), or elamipretide, has been proposed as a first-in-class group of compounds capable of restoring cellular bioenergetics. Elamipretide prevents the conversion of cytochrome c into a peroxidase by interacting with CL, thereby preserving the mitochondrial membrane integrity and enhancing OXPHOS [118,119]. The positive effects of this agent have been demonstrated in models of AD [120,121,122], PD [123], and peripheral mitochondrial injury diseases [124]. A Phase III trial assessing elamipretide in models of mitochondrial myopathy failed to achieve its primary endpoints [125]. One reason is the relatively weak interaction of the SS peptide against CL [126]. Alternatively, a closely related compound to SS-31, known as SBT-272 (bevemipretide trihydrochloride), has been identified. This drug is a novel peptidomimetic under development by Stealth BioTherapeutics Inc. The mechanism of action is similar to SS-31; however, SBT-272 shows a higher mitochondrial uptake, greater concentrations in the brain, and a higher bioavailability [127]. Importantly, SBT-272 counteracts the proteinopathy TDP-43 (TAR DNA-binding protein 43) in ALS upper motor neurons by modulating mitochondrial integrity, motility, and function [127]. SBT-272 is currently in a Phase 1 trial in healthy subjects.
Aside from them, new promising discoveries of CL-targeting peptides have recently been proposed. For instance, the synthetic CMP3013, a cyclohexylalanine-containing α-helical amphipathic peptide, has been demonstrated as a protective agent preserving the mitochondrial cristae structure and enhancing ATP production in models of kidney dysfunction. CMP3013 exhibits high selectivity for CL compared to other IMM lipid components [128]. Remarkably, the pharmacologic inhibition of CL alterations with XJB-5-131, a synthetic mitochondria-targeted electron and reactive oxygen species scavenger, attenuated cell death and tissue damage, and ameliorated motor deficits after SCI in adult rats [93,129]. Notably, supplementation with eicosapentaenoic acid (EPA) from fish oil has been demonstrated to elevate the mitochondrial membrane potential and CL levels in astrocytes. However, it does not alter ATP levels, implying that its utilization may help to prevent neurodegenerative diseases [130]. A recent study also suggests that the bacterial probiotic Weizmannia coagulans lilac-01 (Lilac-01EVs), characterized by a high concentration of CL and PG in the cell membrane, may have the ability to inhibit cell death in primary microglia [131].
Lastly, using computational screening methods, a recent study has discovered a new drug called CardioLipin-Binder (CLiB) that specifically targets CL [132]. This study showed that CLiB increased the respiration of CL-containing intact bacterial cells and isolated mitochondria. Hence, CLiB may serve both basic research and, potentially, therapeutic purposes. Overall, these drugs represent a potential target for pharmacological strategies aimed at treating neurodegeneration. Further research has to be carried out in order to prove their benefits in clinical trials.
7. Conclusions and Perspectives
In the last few years, clinical and experimental studies from human and animal models have demonstrated evidence linking aberrant CL metabolism with neurological dysfunction, suggesting that CL may serve as a biomarker candidate for neurodegenerative diseases. However, the specific physiological roles of CL in different cell types and its contribution to neurodegeneration are not fully understood. Future studies should explore the cell-type-specific aspects of CL biosynthesis and remodeling. Moreover, since brain biopsy samples of living patients are not available, alternative samples such as blood, urine, or saliva should be employed in lipidomic studies to identify “CL signatures” that predict the severity of the disease. Recent discoveries in new drugs and dietary supplements that target CL by regulating its availability in cells should also be studied in human clinical trials for therapeutic purposes. Adopting innovative, multidisciplinary approaches will be essential for a comprehensive understanding of these roles and their impact on nervous system homeostasis and brain function.
Funding
This research was funded by Instituto de Salud Carlos” III, CIBERNED, grant number CB06/05/0041.
Acknowledgments
We want to thank Hector Cordero for the feedback and discussion on the review.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pangborn, M.C. Isolation and Purification of a Serologically Active Phospholipid from Beef Heart. J. Biol. Chem. 1942, 143, 247–256. [Google Scholar] [CrossRef]
- Horvath, S.E.; Daum, G. Lipids of Mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef]
- Beltrán-Heredia, E.; Tsai, F.C.; Salinas-Almaguer, S.; Cao, F.J.; Bassereau, P.; Monroy, F. Membrane Curvature Induces Cardiolipin Sorting. Commun. Biol. 2019, 2, 225. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Itoh, K.; Yamada, T.; Cerveny, K.L.; Suzuki, T.L.; Macdonald, P.; Frohman, M.A.; Ramachandran, R.; Iijima, M.; Sesaki, H. Coincident Phosphatidic Acid Interaction Restrains Drp1 in Mitochondrial Division. Mol. Cell 2016, 63, 1034–1043. [Google Scholar] [CrossRef]
- Taylor, W.A.; Hatch, G.M. Identification of the Human Mitochondrial Linoleoyl-Coenzyme a Monolysocardiolipin Acyltransferase (MLCL AT-1). J. Biol. Chem. 2009, 284, 30360–30371. [Google Scholar] [CrossRef]
- Oemer, G.; Lackner, K.; Muigg, K.; Krumschnabel, G.; Watschinger, K.; Sailer, S.; Lindner, H.; Gnaiger, E.; Wortmann, S.B.; Werner, E.R.; et al. Molecular Structural Diversity of Mitochondrial Cardiolipins. Proc. Natl. Acad. Sci. USA 2018, 115, 4158–4163. [Google Scholar] [CrossRef]
- Oemer, G.; Koch, J.; Wohlfarter, Y.; Alam, M.T.; Lackner, K.; Sailer, S.; Neumann, L.; Lindner, H.H.; Watschinger, K.; Haltmeier, M.; et al. Phospholipid Acyl Chain Diversity Controls the Tissue-Specific Assembly of Mitochondrial Cardiolipins. Cell Rep. 2020, 30, 4281–4291.e4. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria Dysfunction in the Pathogenesis of Alzheimer’s Disease: Recent Advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The Multi-Faceted Role of Mitochondria in the Pathology of Parkinson’s Disease. J. Neurochem. 2020, 156, 715–752. [Google Scholar] [CrossRef]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Büttner, S. Mitochondrial Lipids in Neurodegeneration. Cell Tissue Res. 2017, 367, 125–140. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.M.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin Profile Changes Are Associated to the Early Synaptic Mitochondrial Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Gaudioso, A.; Garcia-Rozas, P.; Casarejos, M.J.; Pastor, O.; Rodriguez-Navarro, J.A. Lipidomic Alterations in the Mitochondria of Aged Parkin Null Mice Relevant to Autophagy. Front. Neurosci. 2019, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Burg, T.; Rossaert, E.; Moisse, M.; Van Damme, P.; Van Den Bosch, L. Histone Deacetylase Inhibition Regulates Lipid Homeostasis in a Mouse Model of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 11224. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Filho, A.B.; Pinto, I.F.D.; Dantas, L.S.; Xavier, A.M.; Inague, A.; Faria, R.L.; Medeiros, M.H.G.; Glezer, I.; Yoshinaga, M.Y.; Miyamoto, S. Alterations in Lipid Metabolism of Spinal Cord Linked to Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 11642. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Hier, Z.E.; Clark, R.S.B.; Kochanek, P.M.; Kagan, V.E.; Bayır, H. Detection of Brain Specific Cardiolipins in Plasma after Experimental Pediatric Head Injury. Exp. Neurol. 2019, 316, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Polimova, A.M.; Maciel, E.; Tyurin, V.A.; Kapralova, V.I.; Winnica, D.E.; Vikulina, A.S.; Domingues, M.R.M.; McCoy, J.; Sanders, L.H.; et al. LC/MS Analysis of Cardiolipins in Substantia Nigra and Plasma of Rotenone-Treated Rats: Implication for Mitochondrial Dysfunction in Parkinson’s Disease. Free Radic. Res. 2015, 49, 681–691. [Google Scholar] [CrossRef]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Lewis, R.N.A.H.; McElhaney, R.N. The Physicochemical Properties of Cardiolipin Bilayers and Cardiolipin-Containing Lipid Membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2069–2079. [Google Scholar] [CrossRef]
- Boyd, K.J.; Alder, N.N.; May, E.R. Buckling under Pressure: Curvature-Based Lipid Segregation and Stability Modulation in Cardiolipin-Containing Bilayers. Langmuir 2017, 33, 6937–6946. [Google Scholar] [CrossRef]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a Mitochondrial Cochaperone Associated with Cardiomyopathy, Forms a Complex with Prohibitins to Regulate Cardiolipin Remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Dimitrijevs, P.; Dimitrijevs, P.; Domracheva, I.; Arsenyan, P. Improved Method for the Preparation of Nonyl Acridine Orange Analogues and Utilization in Detection of Cardiolipin. New J. Chem. 2020, 44, 9626–9633. [Google Scholar] [CrossRef]
- Ryan, T.; Bamm, V.V.; Stykel, M.G.; Coackley, C.L.; Humphries, K.M.; Jamieson-Williams, R.; Ambasudhan, R.; Mosser, D.D.; Lipton, S.A.; Harauz, G.; et al. Cardiolipin Exposure on the Outer Mitochondrial Membrane Modulates α-Synuclein. Nat. Commun. 2018, 9, 817. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.; Cheng, I.F.; Balleininger, M.; Vaz, F.M.; Streckfuss-Bömeke, K.; Hübscher, D.; Vukotic, M.; Wanders, R.J.A.; Rehling, P.; Guan, K. Cardiolipin Deficiency Affects Respiratory Chain Function and Organization in an Induced Pluripotent Stem Cell Model of Barth Syndrome. Stem Cell Res. 2013, 11, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.A.; Ramanathan, A.; Lopez, C.F. Cardiolipin-Dependent Properties of Model Mitochondrial Membranes from Molecular Simulations. Biophys. J. 2019, 117, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.L.; Derreumaux, P.; Nguyen, P.H. Structure and Elasticity of Mitochondrial Membranes: A Molecular Dynamics Simulation Study. J. Phys. Chem. B 2023, 127, 10778–10791. [Google Scholar] [CrossRef] [PubMed]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Cristae Membrane Dynamics—A Paradigm Change. Trends Cell Biol. 2020, 30, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Acehan, D.; Malhotra, A.; Xu, Y.; Ren, M.; Stokes, D.L.; Schlame, M. Cardiolipin Affects the Supramolecular Organization of ATP Synthase in Mitochondria. Biophys. J. 2011, 100, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Mourier, A.; Yamada, J.; Michael McCaffery, J.; Nunnari, J. MICOS Coordinates with Respiratory Complexes and Lipids to Establish Mitochondrial Inner Membrane Architecture. eLife 2015, 2015, e07739. [Google Scholar] [CrossRef]
- Rampelt, H.; Wollweber, F.; Gerke, C.; de Boer, R.; van der Klei, I.J.; Bohnert, M.; Pfanner, N.; van der Laan, M. Assembly of the Mitochondrial Cristae Organizer Mic10 Is Regulated by Mic26–Mic27 Antagonism and Cardiolipin. J. Mol. Biol. 2018, 430, 1883–1890. [Google Scholar] [CrossRef]
- Koob, S.; Barrera, M.; Anand, R.; Reichert, A.S. The Non-Glycosylated Isoform of MIC26 Is a Constituent of the Mammalian MICOS Complex and Promotes Formation of Crista Junctions. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Kondadi, A.K.; Meisterknecht, J.; Golombek, M.; Nortmann, O.; Riedel, J.; Peifer-Weiß, L.; Brocke-Ahmadinejad, N.; Schlütermann, D.; Stork, B.; et al. MIC26 and MIC27 Cooperate to Regulate Cardiolipin Levels and the Landscape of OXPHOS Complexes. Life Sci. Alliance 2020, 3, e202000711. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.L.; Robinson, A.J.; Walker, J.E. Cardiolipin Binds Selectively but Transiently to Conserved Lysine Residues in the Rotor of Metazoan ATP Synthases. Proc. Natl. Acad. Sci. USA 2016, 113, 8687–8692. [Google Scholar] [CrossRef]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the Dimeric ATP Synthase from Bovine Mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.D.; Ball, W.B.; Pryce, E.N.; Gohil, V.M. Specific Requirements of Nonbilayer Phospholipids in Mitochondrial Respiratory Chain Function and Formation. Mol. Biol. Cell 2016, 27, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The Architecture of Respiratory Supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The Multiple Functions of Cytochrome c and Their Regulation in Life and Death Decisions of the Mammalian Cell: From Respiration to Apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Gao, J.; Siira, S.J.; Shearwood, A.M.; Ermer, J.A.; Hofferek, V.; Mathews, J.C.; Zheng, M.; Reid, G.E.; Rackham, O.; et al. Cardiolipin Is Required for Membrane Docking of Mitochondrial Ribosomes and Protein Synthesis. J. Cell Sci. 2020, 133, jcs240374. [Google Scholar] [CrossRef] [PubMed]
- Senoo, N.; Kandasamy, S.; Ogunbona, O.B.; Baile, M.G.; Lu, Y.; Claypool, S.M. Cardiolipin, Conformation, and Respiratory Complex-Dependent Oligomerization of the Major Mitochondrial ADP/ATP Carrier in Yeast. Sci. Adv. 2020, 6, eabb0780. [Google Scholar] [CrossRef]
- Yi, Q.; Yao, S.; Ma, B.; Cang, X. The Effects of Cardiolipin on the Structural Dynamics of the Mitochondrial ADP/ATP Carrier in Its Cytosol-Open State. J. Lipid Res. 2022, 63, 100227. [Google Scholar] [CrossRef]
- Ghosh, S.; Zulkifli, M.; Joshi, A.; Venkatesan, M.; Cristel, A.; Vishnu, N.; Madesh, M.; Gohil, V.M. MCU-Complex-Mediated Mitochondrial Calcium Signaling Is Impaired in Barth Syndrome. Hum. Mol. Genet. 2022, 31, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Ball, W.B.; Madaris, T.R.; Srikantan, S.; Madesh, M.; Mootha, V.K.; Gohil, V.M. An Essential Role for Cardiolipin in the Stability and Function of the Mitochondrial Calcium Uniporter. Proc. Natl. Acad. Sci. USA 2020, 117, 16383–16390. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of Intact Human MCU Supercomplex with the Auxiliary MICU Subunits. Protein Cell 2021, 12, 220–229. [Google Scholar] [CrossRef]
- Li, Y.; Lou, W.; Raja, V.; Denis, S.; Yu, W.; Schmidtke, M.W.; Reynolds, C.A.; Schlame, M.; Houtkooper, R.H.; Greenberg, M.L. Cardiolipin-Induced Activation of Pyruvate Dehydrogenase Links Mitochondrial Lipid Biosynthesis to TCA Cycle Function. J. Biol. Chem. 2019, 294, 11568–11578. [Google Scholar] [CrossRef]
- Li, Y.; Lou, W.; Grevel, A.; Böttinger, L.; Liang, Z.; Ji, J.; Patil, V.A.; Liu, J.; Ye, C.; Hüttemann, M.; et al. Cardiolipin-Deficient Cells Have Decreased Levels of the Iron-Sulfur Biogenesis Protein Frataxin. J. Biol. Chem. 2020, 295, 11928–11937. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, M.; Bharambe, N.; Shang, Y.; Lu, B.; Mandal, A.; Mohan, P.M.; Wang, R.; Boatz, J.C.; Galvez, J.M.M.; Shnyrova, A.V.; et al. NMR Identification of a Conserved Drp1 Cardiolipin-Binding Motif Essential for Stress-Induced Mitochondrial Fission. Proc. Natl. Acad. Sci. USA 2021, 118, e2023079118. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Boopathy, S.; Nguyen, T.H.; Lugo, C.M.; Chao, L.H. Absence of Cardiolipin from the Outer Leaflet of a Mitochondrial Inner Membrane Mimic Restricts Opa1-Mediated Fusion. Front. Mol. Biosci. 2021, 8, 769135. [Google Scholar] [CrossRef] [PubMed]
- von der Malsburg, A.; Sapp, G.M.; Zuccaro, K.E.; von Appen, A.; Moss, F.R.; Kalia, R.; Bennett, J.A.; Abriata, L.A.; Dal Peraro, M.; van der Laan, M.; et al. Structural Mechanism of Mitochondrial Membrane Remodelling by Human OPA1. Nature 2023, 620, 1101–1108. [Google Scholar] [CrossRef]
- Vance, J.E. Inter-Organelle Membrane Contact Sites: Implications for Lipid Metabolism. Biol. Direct 2020, 15, 24. [Google Scholar] [CrossRef]
- Favero, G.; Garcia-Gomez, R.; Monsalve, M.; Rezzani, R.; Lavazza, A.; Stacchiotti, A. Perspective: Mitochondria-ER Contacts in Metabolic Cellular Stress Assessed by Microscopy. Cells 2018, 8, 5. [Google Scholar] [CrossRef]
- Yeo, H.K.; Park, T.H.; Kim, H.Y.; Jang, H.; Lee, J.; Hwang, G.; Ryu, S.E.; Park, S.H.; Song, H.K.; Ban, H.S.; et al. Phospholipid Transfer Function of PTPIP51 at Mitochondria-associated ER Membranes. EMBO Rep. 2021, 22, e51323. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin Remodeling by ALCAT1 Links Oxidative Stress and Mitochondrial Dysfunction to Obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Tirrell, P.S.; Nguyen, K.N.; Luby-Phelps, K.; Friedman, J.R. MICOS Subcomplexes Assemble Independently on the Mitochondrial Inner Membrane in Proximity to ER Contact Sites. J. Cell Biol. 2020, 219, e202003024. [Google Scholar] [CrossRef]
- Iwamoto, M.; Morito, M.; Oiki, S.; Nishitani, Y.; Yamamoto, D.; Matsumori, N. Cardiolipin Binding Enhances KcsA Channel Gating via Both Its Specific and Dianion-Monoanion Interchangeable Sites. iScience 2023, 26, 108471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lyu, J.; Zhu, Y.; Laganowsky, A. Cardiolipin Regulates the Activity of the Mitochondrial ABC Transporter ABCB10. Biochemistry 2023, 62, 3159–3165. [Google Scholar] [CrossRef]
- Liu, G.Y.; Ho Moon, S.; Jenkins, C.M.; Li, M.; Sims, H.F.; Guan, S.; Gross, R.W. The Phospholipase IPLA2 Is a Major Mediator Releasing Oxidized Aliphatic Chains from Cardiolipin, Integrating Mitochondrial Bioenergetics and Signaling. J. Biol. Chem. 2017, 292, 10672–10684. [Google Scholar] [CrossRef]
- Olivar-Villanueva, M.; Ren, M.; Schlame, M.; Phoon, C.K.L. The Critical Role of Cardiolipin in Metazoan Differentiation, Development, and Maturation. Dev. Dyn. 2023, 252, 691–712. [Google Scholar] [CrossRef]
- Schlame, M.; Xu, Y. The Function of Tafazzin, a Mitochondrial Phospholipid–Lysophospholipid Acyltransferase. J. Mol. Biol. 2020, 432, 5043–5051. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I.; Mulkidjanian, A.Y. Cardiolipin, Perhydroxyl Radicals, and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxid. Med. Cell. Longev. 2020, 2020, 1323028. [Google Scholar] [CrossRef]
- Vladimirov, G.K.; Vikulina, A.S.; Volodkin, D.; Vladimirov, Y.A. Structure of the Complex of Cytochrome c with Cardiolipin in Non-Polar Environment. Chem. Phys. Lipids 2018, 214, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E. Peroxidase Activity and Structural Transitions of Cytochrome c Bound to Cardiolipin-Containing Membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A Distinct Pathway Remodels Mitochondrial Cristae and Mobilizes Cytochrome c during Apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Vähäheikkilä, M.; Peltomaa, T.; Róg, T.; Vazdar, M.; Pöyry, S.; Vattulainen, I. How Cardiolipin Peroxidation Alters the Properties of the Inner Mitochondrial Membrane? Chem. Phys. Lipids 2018, 214, 15–23. [Google Scholar] [CrossRef]
- Lacombe, M.L.; Tokarska-Schlattner, M.; Boissan, M.; Schlattner, U. The Mitochondrial Nucleoside Diphosphate Kinase (NDPK-D/NME4), a Moonlighting Protein for Cell Homeostasis. Lab. Investig. 2018, 98, 582–588. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Iriondo, M.N.; Etxaniz, A.; Varela, Y.R.; Ballesteros, U.; Hervás, J.H.; Montes, L.R.; Goñi, F.M.; Alonso, A. LC3 Subfamily in Cardiolipin-Mediated Mitophagy: A Comparison of the LC3A, LC3B and LC3C Homologs. Autophagy 2022, 18, 2985–3003. [Google Scholar] [CrossRef]
- Clifton, L.A.; Wacklin-Knecht, H.P.; Ådén, J.; Ul Mushtaq, A.; Sparrman, T.; Gröbner, G. Creation of Distinctive Bax-Lipid Complexes at Mitochondrial Membrane Surfaces Drives Pore Formation to Initiate Apoptosis. Sci. Adv. 2023, 9, eadg7940. [Google Scholar] [CrossRef]
- Gasanov, S.E.; Kim, A.A.; Yaguzhinsky, L.S.; Dagda, R.K. Non-Bilayer Structures in Mitochondrial Membranes Regulate ATP Synthase Activity. Physiol. Behav. 2016, 176, 139–148. [Google Scholar] [CrossRef]
- Huang, Y.; Powers, C.; Madala, S.K.; Greis, K.D.; Haffey, W.D.; Towbin, J.A.; Purevjav, E.; Javadov, S.; Strauss, A.W.; Khuchua, Z. Cardiac Metabolic Pathways Affected in the Mouse Model of Barth Syndrome. PLoS ONE 2015, 10, e0128561. [Google Scholar] [CrossRef]
- Anzmann, A.F.; Sniezek, O.L.; Pado, A.; Busa, V.; Vaz, F.M.; Kreimer, S.D.; DeVine, L.R.; Cole, R.N.; Le, A.; Kirsch, B.J.; et al. Diverse Mitochondrial Abnormalities in a New Cellular Model of TAFFAZZIN Deficiency Are Remediated by Cardiolipin-Interacting Small Molecules. J. Biol. Chem. 2021, 297, 101005. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Hatch, A.L.; Higgs, H.N. Effects of Phosphorylation on Drp1 Activation by Its Receptors, Actin, and Cardiolipin. Prepint 2023, 35, ar16. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome C Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Amoscato, A.A.; Sparvero, L.J.; He, R.R.; Watkins, S.; Bayir, H.; Kagan, V.E. Imaging Mass Spectrometry of Diversified Cardiolipin Molecular Species in the Brain. Anal. Chem. 2014, 86, 6587–6595. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Kline, A.E.; Amoscato, A.; Arias, A.S.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Global Lipidomics Identifies Cardiolipin Oxidation as a Mitochondrial Target for Redox Therapy of Acute Brain Injury. Physiol. Behav. 2012, 176, 139–148. [Google Scholar]
- Schlame, M.; Brody, S.; Hostetler, K.Y. Mitochondrial Cardiolipin in Diverse Eukaryotes: Comparison of Biosynthetic Reactions and Molecular Acyl Species. Eur. J. Biochem. 1993, 212, 727–733. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Amoscato, A.A.; Fink, A.B.; Anthonymuthu, T.; New, L.A.; Kochanek, P.M.; Watkins, S.; Kagan, V.E.; Bayır, H. Imaging Mass Spectrometry Reveals Loss of Polyunsaturated Cardiolipins in the Cortical Contusion, Hippocampus, and Thalamus after Traumatic Brain Injury. J. Neurochem. 2016, 139, 659–675. [Google Scholar] [CrossRef]
- Kolomiytseva, I.K.; Markevich, L.N.; Ignat’Ev, D.A.; Bykova, O.V. Lipids of Nuclear Fractions from Neurons and Glia of Rat Neocortex under Conditions of Artificial Hypobiosis. Biochemistry 2010, 75, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, F.M.; Cafagna, F.; Petruzzella, V.; Gadaleta, M.N.; Quagliariello, E. Lipid Composition in Synaptic and Nonsynaptic Mitochondria from Rat Brains and Effect of Aging. J. Neurochem. 1992, 59, 487–491. [Google Scholar] [CrossRef]
- Kurokin, I.; Lauer, A.A.; Janitschke, D.; Winkler, J.; Theiss, E.L.; Griebsch, L.V.; Pilz, S.M.; Matschke, V.; van der Laan, M.; Grimm, H.S.; et al. Targeted Lipidomics of Mitochondria in a Cellular Alzheimer’s Disease Model. Biomedicines 2021, 9, 81. [Google Scholar] [CrossRef]
- Guan, Z.; Wang, Y.; Cairns, N.J.; Lantos, P.L.; Dallner, G.; Sindelar, P.J. Decrease and Structural Modifications of Phosphatidylethanolamine Plasmalogen in the Brain with Alzheimer Disease. J. Neurophatology Exp. Neurol. 1994, 58, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, A.; Ghio, S.; Caruana, M.; Weckbecker, D.; Schmidt, F.; Kamp, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Giese, A.; et al. Tau-Induced Mitochondrial Membrane Perturbation Is Dependent upon Cardiolipin. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183064. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.J.; Ranger, A.L.; Murray, T.; McRae, S.; Klegeris, A. Extracellular Cardiolipin Modulates Glial Phagocytosis and Cytokine Secretion in a Toll-like Receptor 4-dependent Manner. Alzheimer’s Dement. 2020, 16, e047338. [Google Scholar] [CrossRef]
- Kawatani, K.; Holm, M.L.; Starling, S.C.; Martens, Y.A.; Zhao, J.; Lu, W.; Ren, Y.; Li, Z.; Jiang, P.; Jiang, Y.; et al. ABCA7 Deficiency Causes Neuronal Dysregulation by Altering Mitochondrial Lipid Metabolism. Mol. Psychiatry 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.L.; Annesley, S.J.; Gordon, S.E.; Mroczek, K.; Perugini, M.A.; Lawson, V.A.; Fisher, P.R.; Finkelstein, D.I.; Hill, A.F. Misfolded α-Synuclein Causes Hyperactive Respiration without Functional Deficit in Live Neuroblastoma Cells. DMM Dis. Model. Mech. 2020, 13, dmm040899. [Google Scholar] [CrossRef] [PubMed]
- Ghio, S.; Camilleri, A.; Caruana, M.; Ruf, V.C.; Schmidt, F.; Leonov, A.; Ryazanov, S.; Griesinger, C.; Cauchi, R.J.; Kamp, F.; et al. Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. ACS Chem. Neurosci. 2019, 10, 3815–3829. [Google Scholar] [CrossRef] [PubMed]
- Zhaliazka, K.; Ali, A.; Kurouski, D. Phospholipids and Cholesterol Determine Molecular Mechanisms of Cytotoxicity of α-Synuclein Oligomers and Fibrils. ACS Chem. Neurosci. 2023, 15, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zhang, J.; Qi, S.; Liu, Z.; Zhang, X.; Zheng, Y.; Andersen, J.P.; Zhang, W.; Strong, R.; Martinez, P.A.; et al. Cardiolipin Remodeling by ALCAT1 Links Mitochondrial Dysfunction to Parkinson’s Diseases. Aging Cell 2019, 18, e12941. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Choi, H.; Chevalier, A.; Hogan, D.; Akgoc, Z.; Schneider, J.S. Sex-Related Abnormalities in Substantia Nigra Lipids in Parkinson’s Disease. ASN Neuro 2018, 10, 1759091418781889. [Google Scholar] [CrossRef]
- Phan, K.; He, Y.; Bhatia, S.; Pickford, R.; Mcdonald, G.; Mazumder, S.; Timmins, H.C.; Hodges, J.R.; Piguet, O.; Dzamko, N.; et al. Multiple Pathways of Lipid Dysregulation in Amyotrophic Lateral Sclerosis. Brain Commun. 2023, 5, fcac340. [Google Scholar] [CrossRef]
- Giudetti, A.M.; Guerra, F.; Longo, S.; Beli, R.; Romano, R.; Manganelli, F.; Nolano, M.; Mangini, V.; Santoro, L.; Bucci, C. An Altered Lipid Metabolism Characterizes Charcot-Marie-Tooth Type 2B Peripheral Neuropathy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158805. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.; Lin, C.; Zuo, Q.; Liu, Y.; Xiao, M.; Xu, X.; Li, Z.; Bao, Z.; Chen, H.; You, Y.; et al. Cardiolipin-Dependent Mitophagy Guides Outcome after Traumatic Brain Injury. J. Neurosci. 2019, 39, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.K.; Deng, L.X.; Wang, M.; Lu, Q.B.; Wang, C.; Wu, X.; Wu, W.; Wang, Y.; Qu, W.; Han, Q.; et al. Restoring Mitochondrial Cardiolipin Homeostasis Reduces Cell Death and Promotes Recovery after Spinal Cord Injury. Cell Death Dis. 2022, 13, 1058. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Alzheimer’s Disease. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and Controversies Surrounding the Amyloid Hypothesis of Alzheimer’s Disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated Function of Mitochondria-Associated ER Membranes in Alzheimer Disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Yin, F. Lipid Metabolism and Alzheimer’s Disease: Clinical Evidence, Mechanistic Link and Therapeutic Promise. FEBS J. 2023, 290, 1420–1453. [Google Scholar] [CrossRef] [PubMed]
- Heverin, M.; Bogdanovic, N.; Lütjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Björkhem, I. Changes in the Levels of Cerebral and Extracerebral Sterols in the Brain of Patients with Alzheimer’s Disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human Wild-Type Full-Length Tau Accumulation Disrupts Mitochondrial Dynamics and the Functions via Increasing Mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Tiberi, M.; Matteocci, A.; Fazio, F.; Siffeti, H.; Saracini, S.; Mercuri, N.B.; Sancesario, G. Lipidomics of Bioactive Lipids in Alzheimer’s and Parkinson’s Diseases: Where Are We? Int. J. Mol. Sci. 2022, 23, 6235. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, A.; Debbabi, M.; Bezine, M.; Karym, E.M.; Badred, A. Lipid Biomarkers in Alzheimer’s Disease Lipid Biomarkers in Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 14, 303–312. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. The α-Synucleinopathies: Parkinson’s Disease, Dementia with Lewy Bodies, and Multiple System Atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein Misfolding and Aggregation: Implications in Parkinson’s Disease Pathogenesis. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Gilmozzi, V.; Gentile, G.; Paulina Castelo Rueda, M.; Hicks, A.A.; Pramstaller, P.P.; Zanon, A.; Lévesque, M.; Pichler, I. Interaction of Alpha-Synuclein with Lipids: Mitochondrial Cardiolipin as a Critical Player in the Pathogenesis of Parkinson’s Disease. Front. Neurosci. 2020, 14, 578993. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Kapralov, A.A.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Belikova, N.A.; Vlasova, I.I.; Maeda, A.; et al. Peroxidase Mechanism of Lipid-Dependent Cross-Linking of Synuclein with Cytochrome c. Protection against Apoptosis versus Delayed Oxidative Stress in Parkinson Disease. J. Biol. Chem. 2009, 284, 15951–15969. [Google Scholar] [CrossRef]
- Plotegher, N.; Gratton, E.; Bubacco, L. Number and Brightness Analysis of Alpha-Synuclein Oligomerization and the Associated Mitochondrial Morphology Alterations in Live Cells. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2014–2024. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Erustes, A.G.; Guarache, G.C.; Guedes, E.D.C.; Leão, A.H.F.F.; Pereira, G.J.D.S.; Smaili, S.S. α-Synuclein Interactions in Mitochondria-ER Contacts: A Possible Role in Parkinson’s Disease. Contact 2022, 5, 25152564221119347. [Google Scholar] [CrossRef]
- Avisar, H.; Guardia-Laguarta, C.; Area-Gomez, E.; Surface, M.; Chan, A.K.; Alcalay, R.N.; Lerner, B. Lipidomics Prediction of Parkinson’s Disease Severity: A Machine-Learning Analysis. J. Parkinsons. Dis. 2021, 11, 1141–1155. [Google Scholar] [CrossRef]
- Zardini Buzatto, A.; Tatlay, J.; Bajwa, B.; Mung, D.; Camicioli, R.; Dixon, R.A.; Li, L. Comprehensive Serum Lipidomics for Detecting Incipient Dementia in Parkinson’s Disease. J. Proteome Res. 2021, 20, 4053–4067. [Google Scholar] [CrossRef]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated Analyses of Microbiome and Longitudinal Metabolome Data Reveal Microbial-Host Interactions on Sulfur Metabolism in Parkinson’s Disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Robert, H.; Brown, J. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 108, 37–44. [Google Scholar] [CrossRef]
- Agrawal, I.; Lim, Y.S.; Ng, S.Y.; Ling, S.C. Deciphering Lipid Dysregulation in ALS: From Mechanisms to Translational Medicine. Transl. Neurodegener. 2022, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef]
- Cherry, S.; Jin, E.J.; Özel, M.N.; Lu, Z.; Agi, E.; Wang, D.; Jung, W.-H.; Epstein, D.; Meinertzhagen, I.A.; Chan, C.-C.; et al. Charcot-Marie-Tooth 2B Mutations in Rab7 Cause Dosage-Dependent Neurodegeneration Due to Partial Loss of Function. Elife 2013, 2, e01064. [Google Scholar] [CrossRef]
- Harris, G.; Stickland, C.A.; Lim, M.; Goldberg Oppenheimer, P. Raman Spectroscopy Spectral Fingerprints of Biomarkers of Traumatic Brain Injury. Cells 2023, 12, e01064. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-Class Cardiolipin-Protective Compound as a Therapeutic Agent to Restore Mitochondrial Bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Chavez, J.D.; Tang, X.; Campbell, M.D.; Reyes, G.; Kramer, P.A.; Stuppard, R.; Keller, A.; Zhang, H.; Rabinovitch, P.S.; Marcinek, D.J.; et al. Mitochondrial Protein Interaction Landscape of SS-31. Proc. Natl. Acad. Sci. USA 2020, 117, 15363–15373. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, Z.; Cao, J.; Fu, Q.; Wu, Y.; Zhang, X.; Long, Y.; Zhang, X.; Yang, Y.; Li, Y.; et al. Elamipretide (SS-31) Improves Mitochondrial Dysfunction, Synaptic and Memory Impairment Induced by Lipopolysaccharide in Mice. J. Neuroinflammation 2019, 16, 230. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired Mitochondrial Biogenesis, Defective Axonal Transport of Mitochondria, Abnormal Mitochondrial Dynamics and Synaptic Degeneration in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Manczak, M.; Yin, X.; Arubala, P.; States, U.; Campus, S.W.; States, U.; States, U.; States, U.; States, U.; et al. Synergistic Protective Effects of Mitochondrial Division Inhibitor 1 and Mitochondria-Targeted Small Peptide SS31 in Alzheimer’s Disease P. J. Alzheimers Dis. 2018, 62, 1549–1565. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria Targeted Peptides Protect Against. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The Cardiolipin-Binding Peptide Elamipretide Mitigates Fragmentation of Cristae Networks Following Cardiac Ischemia Reperfusion in Rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef]
- Karaa, A.; Bertini, E.; Carelli, V.; Cohen, B.H.; Enns, G.M.; Falk, M.J.; Goldstein, A.; Gorman, G.S.; Haas, R.; Hirano, M.; et al. Efficacy and Safety of Elamipretide in Individuals with Primary Mitochondrial Myopathy: The MMPOWER-3 Randomized Clinical Trial. Neurology 2023, 101, e238–e252. [Google Scholar] [CrossRef]
- Mitchell, W.; Ng, E.A.; Tamucci, J.D.; Boyd, K.J.; Sathappa, M.; Coscia, A.; Pan, M.; Han, X.; Eddy, N.A.; May, E.R.; et al. The Mitochondria-Targeted Peptide SS-31 Binds Lipid Bilayers and Modulates Surface Electrostatics as a Key Component of Its Mechanism of Action. J. Biol. Chem. 2020, 295, 7452–7469. [Google Scholar] [CrossRef]
- Gautam, M.; Genç, B.; Helmold, B.; Ahrens, A.; Kuka, J.; Makrecka-Kuka, M.; Günay, A.; Koçak, N.; Aguilar-Wickings, I.R.; Keefe, D.; et al. SBT-272 Improves TDP-43 Pathology in ALS Upper Motor Neurons by Modulating Mitochondrial Integrity, Motility, and Function. Neurobiol. Dis. 2023, 178, 106022. [Google Scholar] [CrossRef]
- Shin, G.; Hyun, S.; Kim, D.; Choi, Y.; Kim, K.H.; Kim, D.; Kwon, S.; Kim, Y.S.; Yang, S.H.; Yu, J. Cyclohexylalanine-Containing α-Helical Amphipathic Peptide Targets Cardiolipin, Rescuing Mitochondrial Dysfunction in Kidney Injury. J. Med. Chem. 2023, 67, 3385–3399. [Google Scholar] [CrossRef]
- Xun, Z.; Wipf, P.; McMurray, C.T. XJB-5-131 Is a Mild Uncoupler of Oxidative Phosphorylation. J. Huntingtons. Dis. 2022, 11, 141–151. [Google Scholar] [CrossRef]
- Stulczewski, D.; Zgorzynska, E.; Dziedzic, B.; Wieczorek-Szukala, K.; Szafraniec, K.; Walczewska, A. EPA Stronger than DHA Increases the Mitochondrial Membrane Potential and Cardiolipin Levels but Does Not Change the ATP Level in Astrocytes. Exp. Cell Res. 2023, 424, 113491. [Google Scholar] [CrossRef]
- Minamida, K.; Taira, T.; Sasaki, M.; Higuchi, O.; Meng, X.-Y.; Kamagata, Y.; Miwa, K. Extracellular Vesicles of Weizmannia Coagulans Lilac-01 Reduced Cell Death of Primary Microglia and Increased Mitochondrial Content in Dermal Fibroblasts in Vitro. Biosci. Biotechnol. Biochem. 2023, 88, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Kleinwächter, I.; Mohr, B.; Joppe, A.; Hellmann, N.; Bereau, T.; Osiewacz, H.D.; Schneider, D. CLiB—A Novel Cardiolipin-Binder Isolated via Data-Driven and in Vitro Screening. RSC Chem. Biol. 2022, 3, 941–954. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of CL biosynthesis and remodeling in mammals. CL production begins with PA, which can be sourced from several pathways where the enzymes GTATs and/or mitoPLD are present. PA needs to be transported to the IMM, where CL biosynthetic enzymes reside. PA shuttling from the OMM to the IMM is performed by PRELID1-TRIAP1. Inside of the IMM, PA is converted to CLn in a series of reactions catalyzed by several enzymes. Finally, CLn suffers a remodeling to become CLm in the IMM or OMM. Figure created with BioRender.com (accessed on 1 March 2024). Abbreviations: G3P, glycerol-3-phosphate, GPATs, G3P acyltransferases, AGPATs, LPA acyltransferases; PA, phosphatidic acid; CL, cardiolipin; PRELID1, protein of relevant evolutionary and lymphoid interest domain; TRIAP1, TP53-regulated inhibitor of apoptosis 1; TAM41, translocator assembly and maintenance homolog; PGS1, phosphatidylglycerol phosphate synthase; PTPMT1, protein-tyrosine phosphatase mitochondrial 1; CLS1, CL synthase; CLn, nascent CL; CLm, mature CL, iPLA2, phospholipase A2; MLCL, monolysocardiolipin; TAFAZZIN, tafazzin phospholipid remodeling enzyme; ALCAT1, acyl-CoA:lysocardiolipin acyltransferase 1; MLCLAT1, acyl-CoA:monolysocardiolipin acyltransferase 1.
Figure 1.
Schematic representation of CL biosynthesis and remodeling in mammals. CL production begins with PA, which can be sourced from several pathways where the enzymes GTATs and/or mitoPLD are present. PA needs to be transported to the IMM, where CL biosynthetic enzymes reside. PA shuttling from the OMM to the IMM is performed by PRELID1-TRIAP1. Inside of the IMM, PA is converted to CLn in a series of reactions catalyzed by several enzymes. Finally, CLn suffers a remodeling to become CLm in the IMM or OMM. Figure created with BioRender.com (accessed on 1 March 2024). Abbreviations: G3P, glycerol-3-phosphate, GPATs, G3P acyltransferases, AGPATs, LPA acyltransferases; PA, phosphatidic acid; CL, cardiolipin; PRELID1, protein of relevant evolutionary and lymphoid interest domain; TRIAP1, TP53-regulated inhibitor of apoptosis 1; TAM41, translocator assembly and maintenance homolog; PGS1, phosphatidylglycerol phosphate synthase; PTPMT1, protein-tyrosine phosphatase mitochondrial 1; CLS1, CL synthase; CLn, nascent CL; CLm, mature CL, iPLA2, phospholipase A2; MLCL, monolysocardiolipin; TAFAZZIN, tafazzin phospholipid remodeling enzyme; ALCAT1, acyl-CoA:lysocardiolipin acyltransferase 1; MLCLAT1, acyl-CoA:monolysocardiolipin acyltransferase 1.

Figure 2.
Schematic representation of CL sources (tissues, cells, and fluids), CL structure, and techniques for its detection.
Figure 2.
Schematic representation of CL sources (tissues, cells, and fluids), CL structure, and techniques for its detection.

Figure 3.
Schematic representation of proteins and biological processes affected by CL alterations within mitochondria. Abbreviations: AAC, ADP/ATP carrier; I, III, IV, complex I, III, IV (respectively); MUC, mitochondrial Ca2+ uniporter; MICU1, mitochondrial calcium uptake 1; OPA1, OXPHOS, oxidative phosphorylation; PARL, presenilin-associated rhomboid-like protein; DRP1; dynamin-related protein 1; OPA1, optic atrophy 1; LC3, microtubule-associated protein 1A/1B-light chain 3; Cyt c, cytochrome c; MIC10, mitochondrial contact site and cristae organizing system 10.
Figure 3.
Schematic representation of proteins and biological processes affected by CL alterations within mitochondria. Abbreviations: AAC, ADP/ATP carrier; I, III, IV, complex I, III, IV (respectively); MUC, mitochondrial Ca2+ uniporter; MICU1, mitochondrial calcium uptake 1; OPA1, OXPHOS, oxidative phosphorylation; PARL, presenilin-associated rhomboid-like protein; DRP1; dynamin-related protein 1; OPA1, optic atrophy 1; LC3, microtubule-associated protein 1A/1B-light chain 3; Cyt c, cytochrome c; MIC10, mitochondrial contact site and cristae organizing system 10.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of proteins affected by CL dysfunction.
| Protein Affected | Mitochondrial Alterations | Biological Sample | Ref. |
|---|---|---|---|
| MIC10 subunit | -Cristae defects -Misdistribution of MRC | -Yeast -Hela, HepG2 cells | [29,30,31] |
| F1Fo ATP synthase | -Non-bilayer structure formation -Decrease dimerization ATP synthase in the cristae -Reduce OXPHOS | -Synthetic liposomes -Drosophila | [28,69] |
| -RCs (I, III) -RSCs (I, III, IV) | -Reduction in RCs -Misfold of RSCs | -Mouse | [70] |
| ADP/ATP carrier (AAC) | -Destabilized protein structure -OXPHOS defects. | -Yeast -Human cell lines | [39] |
| MCU, MICU1 | -Reduced mitochondrial Ca2+ uptake -Inactivation of pyruvate dehydrogenase | -BTHS patient cells and cardiac tissue | [41,42] |
| PARL | -Increase the expression of PARL leading to apoptosis | HEK293 cells | [71] |
| DRP1 | -Reduce mitochondrial fission | -Synthetic liposomes | [47] |
| p-ser579-DRP1 p-ser600-DRP1 | -Reduce DRP1 activation | -Synthetic liposomes | [72] |
| OPA1 | -Alterations mitochondrial fusion | -Synthetic liposomes | [48] |
| LC3 | -Decrease mitophagy | -Synthetic liposomes -SH-SY5Y cells | [67] |
| Cytochrome c | -Apoptosis | -HL-60 cells | [73] |
Abbreviations: BTHS, Barth syndrome; CL, cardiolipin; MIC, mitochondrial contact site and cristae organizing system; MUC, mitochondrial calcium uniporter; MICU, mitochondrial calcium uptake 1; RSCs; respiratory supercomplexes; RCs respiratory complexes; OXPHOS, oxidative phosphorylation; PARL, presenilin-associated rhomboid-like protein; DRP1; dynamin-related protein 1; OPA1, optic atrophy 1; LC3, microtubule-associated protein 1A/1B-light chain 3.
Table 2.
Recent findings in CL-associated neurodegenerative diseases.
| Disease | Model | Findings | References |
|---|---|---|---|
| AD | Brain from 3xTg-AD mice | -Reduced CL species in synaptic mitochondria -Lack of detection of oxidated CL | [11] |
| SH-SY5Y-APPswedish | -Decrease in total CL content -Alterations in PG | [80] | |
| Brain from AD patients | -Slightly reduction of CL content -Decrease in FA content | [81] | |
| SH-SY5Y | -Tau protein exhibits a preference for binding to CL-rich regions of the OMM | [82] | |
| Primary microglia cultures and neuron cell lines | -CL inhibits amyloid-β secretion | [83] | |
| -Human iPSC ABCA7-KO -Brain from Abca7-KO mice | -Reduction of CL content -Decrease ATP synthesis, increase in ROS, and increase mitochondrial fusion | [84] | |
| PD | SH-SY5Y | -CL accelerates the rate of α-synuclein fibrillization, leading to hyperactive respiration | [85] |
| SNCA-mutant human pluripotent stem cells (iPSCs) and SNCA-transgenic mice | -Exposed CL to the OMM binds to and facilitates refolding of α-syn fibril. -Prolonged CL exposure in SNCA-mutants initiates recruitment of LC3 to the mitochondria and mitophagy. | [23] | |
| Freshly isolated mitochondria or liposome | -CL interacts with α-syn to favor pore formation within mitochondrial membranes | [86] | |
| N27 rat dopaminergic cell line | -CL increase α-synuclein aggregation, leading to ER stress | [87] | |
| Brain from MPTP mouse | -Inhibition of ALCAT1 prevents neurotoxicity, apoptosis, and motor deficiencies. | [88] | |
| Brain from Parkin-KO mice | -Lack of changes in CL content -CL remodeling defects with increase of short, saturated CL acyl-chains | [12,89] | |
| Brain from rat rotenone model | -Exposure to rotenone induces a loss in linoleic acid-containing CL species and an increase in CL oxidation | [16] | |
| ALS | Spinal cord from FUS mice | -Reduction in CL content | [13] |
| Cortex and spinal cord from SOD1-G86R mouse | -Reduction in CL content | [14] | |
| Serum ALS patients | -No changes in CL content -Increase in unsaturated lipids | [90] | |
| CMT2B | Fibroblasts from CMT2B patient | -Lack of measure of CL content by lipidomics -Increase in levels of unsaturated FA | [91] |
| TBI | Brain from TBI rat model | -Reduction of CL content | [77] |
| Brain from TBI patients and TBI rat models | -CL drives mitophagy | [92] | |
| SCI | Spine from SCI rat models | -Decrease in CL content and increase in CL oxidation | [93] |
Abbreviations: CL, cardiolipin; Tg, transgenic, AD, Alzheimer’s disease; APP, amyloid precursor protein; PG, phosphatidylglycerol; FA, fatty acids; OMM, outer mitochondrial membrane; iPSC, induced pluripotent stem cells; ABCA ATP-binding cassette sub-family A member; ROS, reactive oxygen species; PD, Parkinson’s disease, SNCA, synuclein alpha; KO, knockout; LC3, microtubule-associated protein 1A/1B-light chain 3; ER, endoplasmic reticulum; MPTP, methyl-4-phenyl-1,2,3,6-tetrahydropyridine; ALCAT1, acyl-CoA:lysocardiolipin acyltransferase-1; ALS, amyotrophic lateral sclerosis; FUS, FUS RNA-binding protein; SOD, superoxide dismutase 1; CMT2B, Charcot–Marie–Tooth Type 2B; TBI, traumatic brain injury; SCI, spinal cord injury.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fuentes, J.M.; Morcillo, P. The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases. Cells 2024, 13, 609. https://0-doi-org.brum.beds.ac.uk/10.3390/cells13070609
AMA Style
Fuentes JM, Morcillo P. The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases. Cells. 2024; 13(7):609. https://0-doi-org.brum.beds.ac.uk/10.3390/cells13070609
Chicago/Turabian StyleFuentes, José M., and Patricia Morcillo. 2024. "The Role of Cardiolipin in Mitochondrial Function and Neurodegenerative Diseases" Cells 13, no. 7: 609. https://0-doi-org.brum.beds.ac.uk/10.3390/cells13070609
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.