A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1


Abstract
:
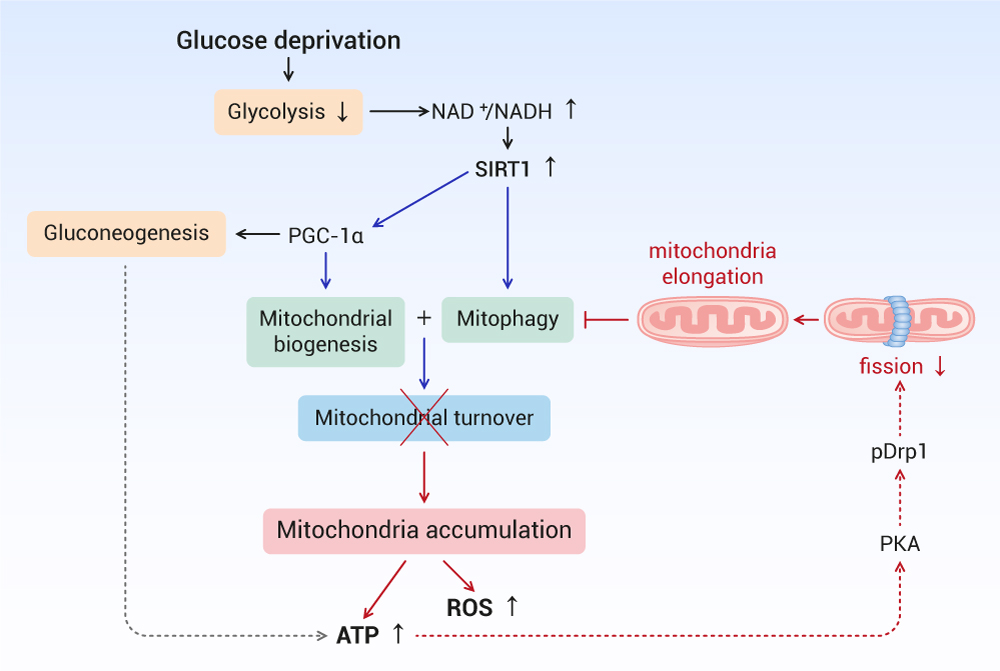{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemical Treatments
2.2. ATP Measurement
2.3. Measurements of OCR and ECAR
2.4. siRNA Transfection
2.5. Western Blot Analysis
2.6. Confocal Microscopy
2.7. Flow Cytometry
2.8. Quantitative PCR
2.9. Measurement of NAD+/NADH
2.10. Statistical Analysis
3. Results
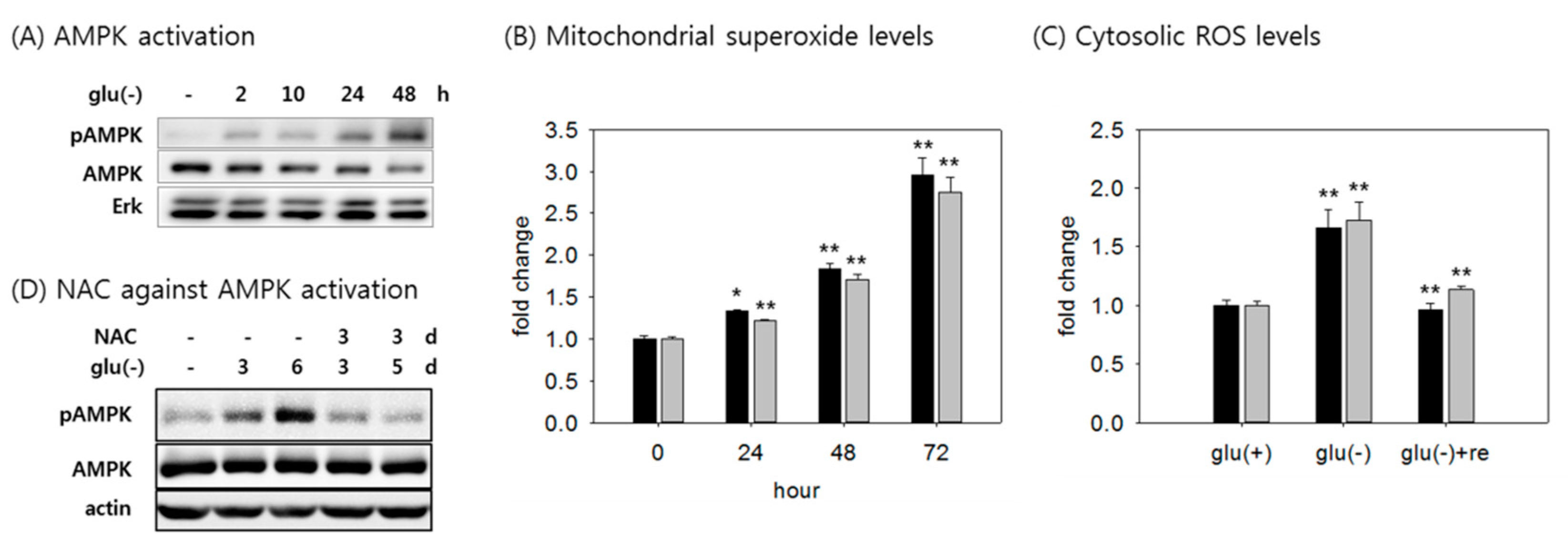
3.1. Cellular ATP Level is Enhanced Upon Glucose Withdrawal Through Increased Mitochondrial ATP Production
3.2. AMPK is Activated by Increased ROS Level
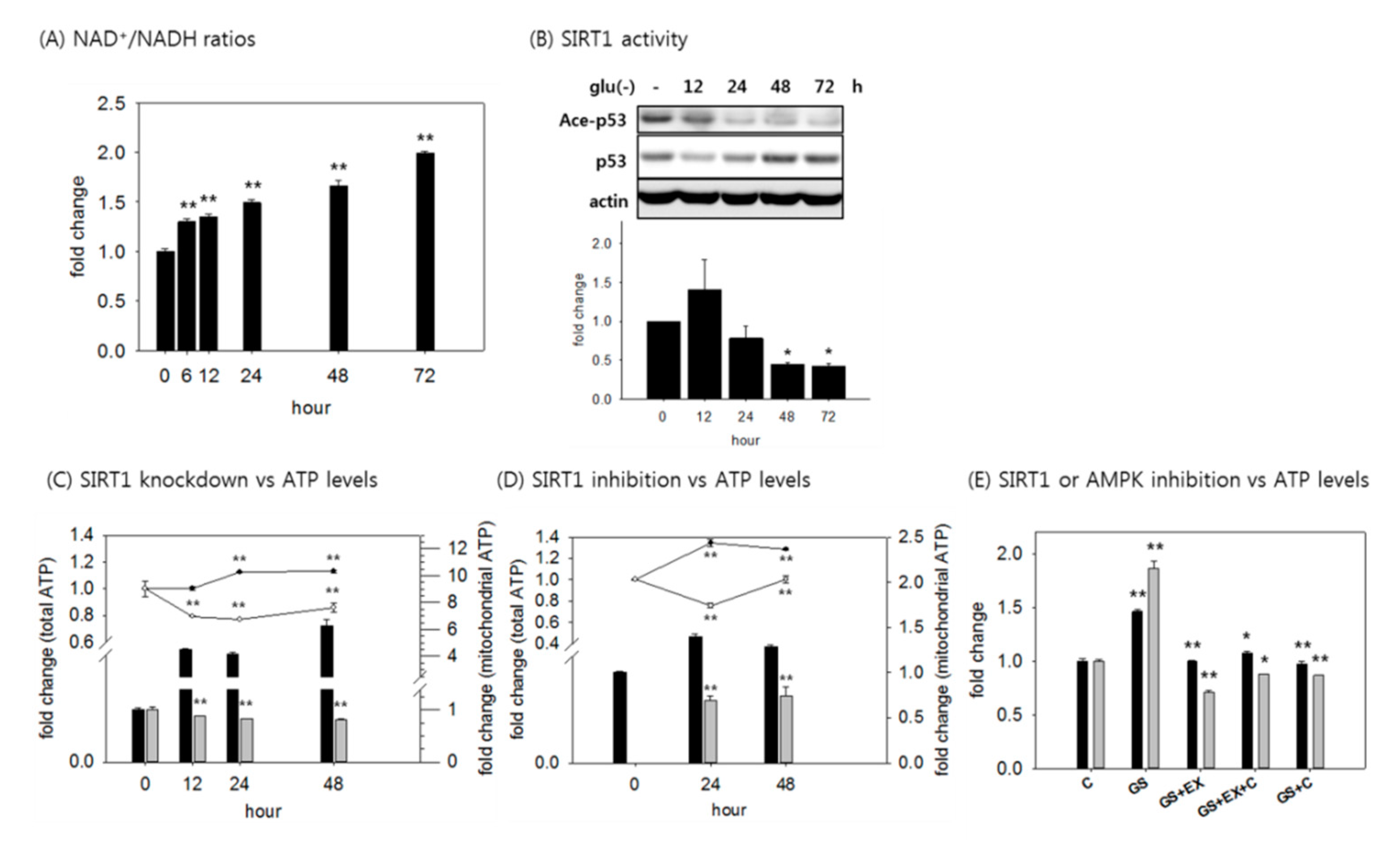
3.3. An Increase in Mitochondrial ATP Production is Mediated by Activated SIRT1 and AMPK
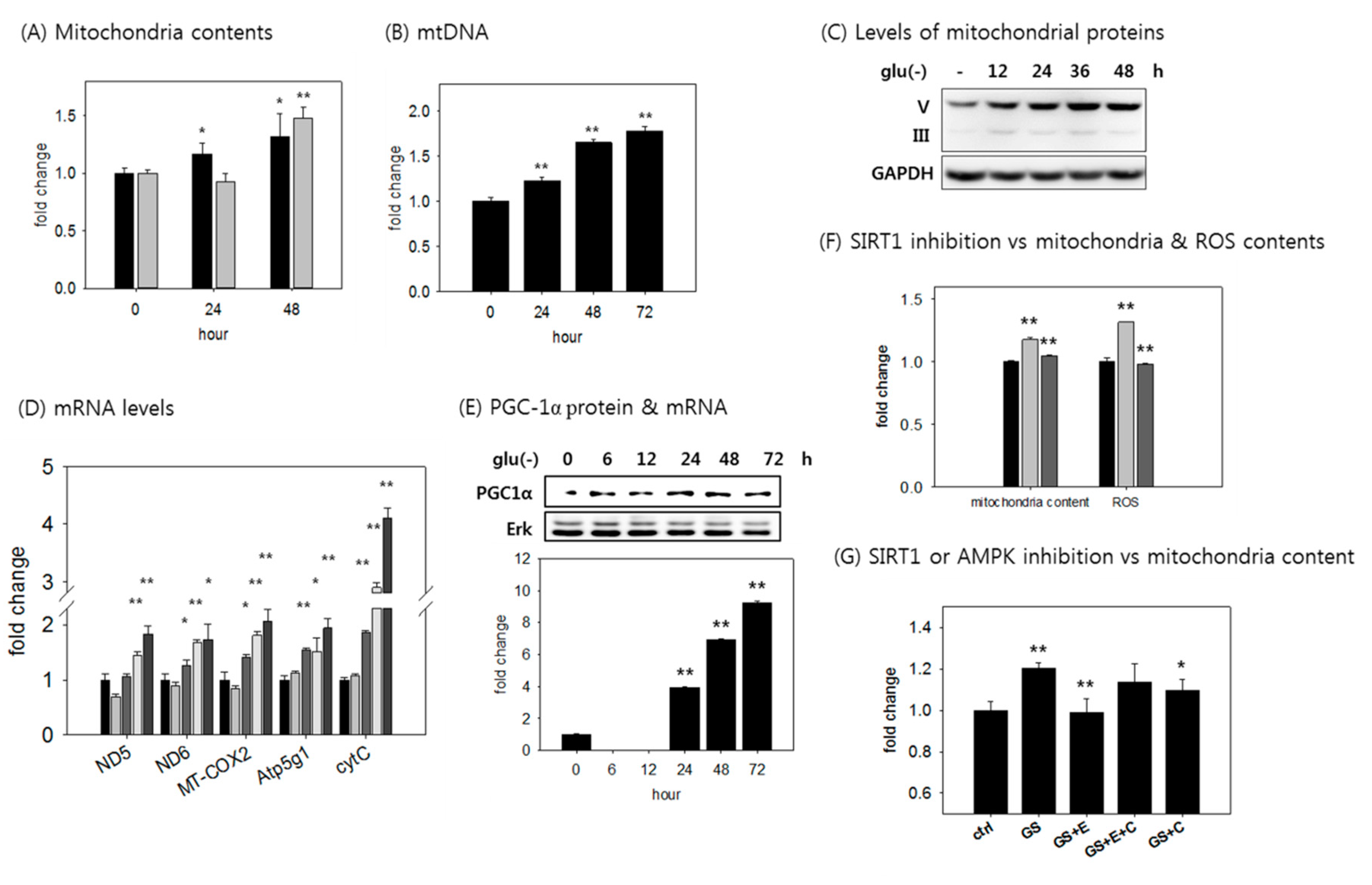
3.4. SIRT1 Activation is Responsible for the Increased ATP Level in Glucose-Deprived Cells Through Enhancing Mitochondrial Biogenesis and Glycolysis
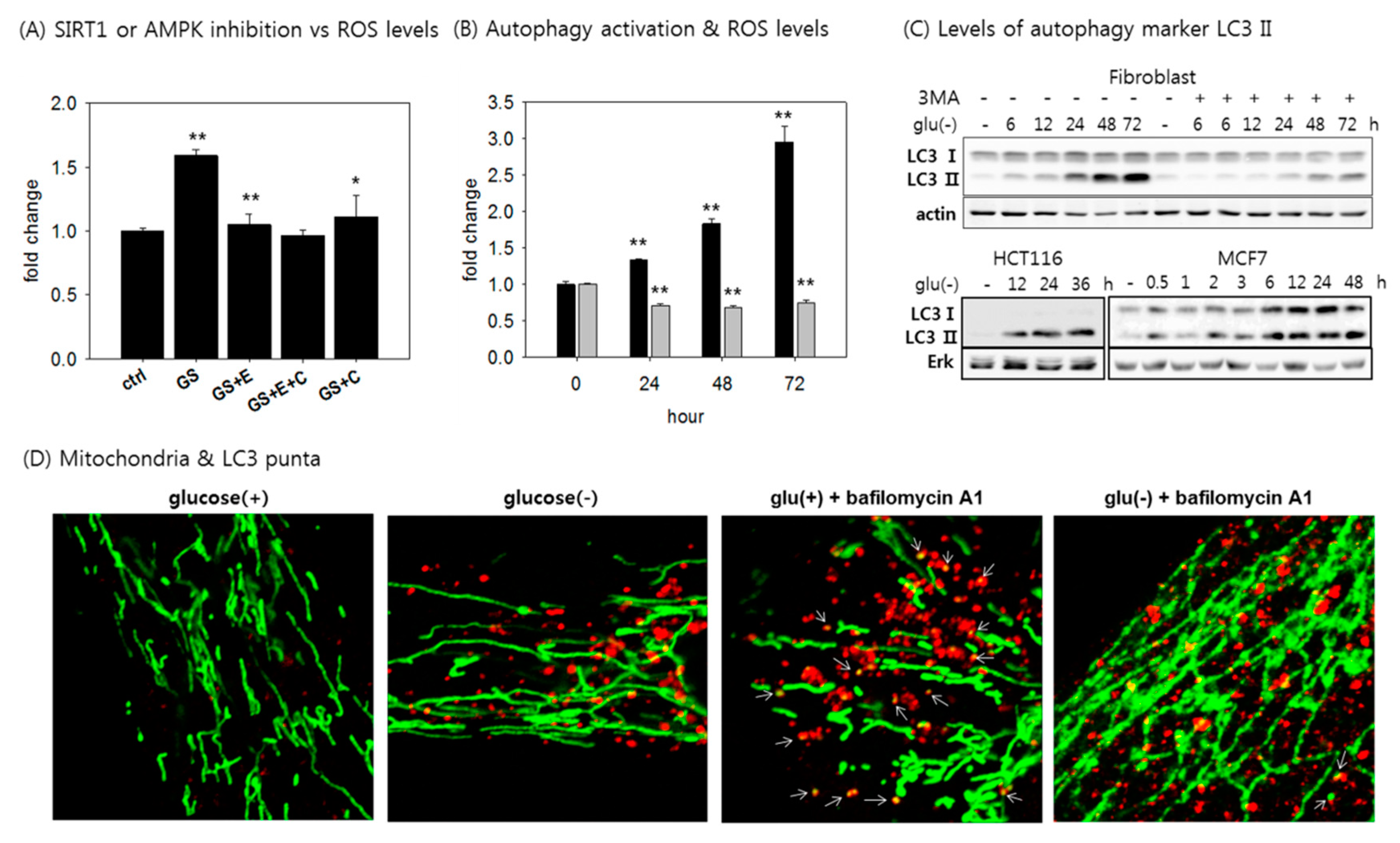
3.5. Autophagy is Activated, but Mitophagy is Attenuated in Glucose-Deprived Cells.
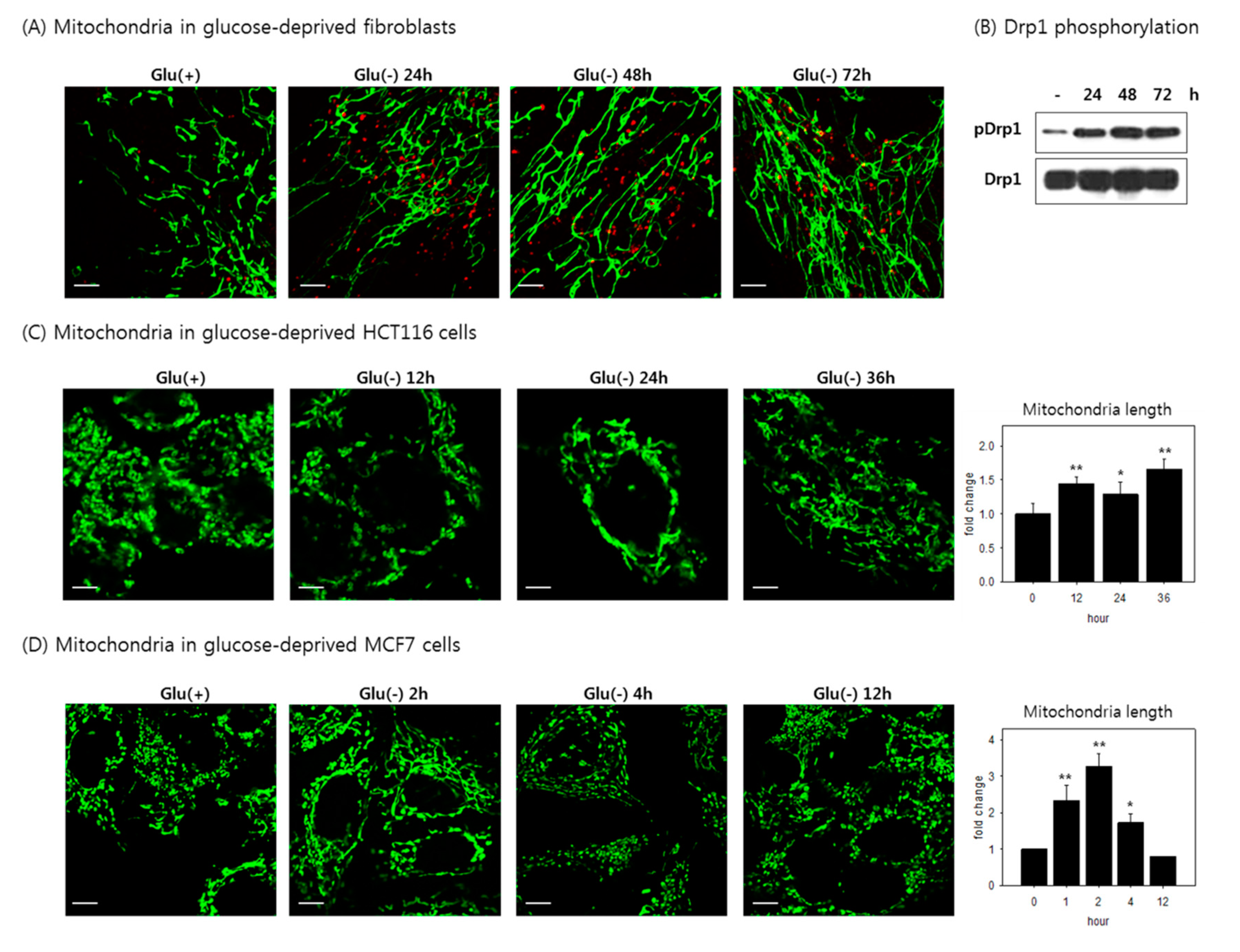
3.6. Mitochondria Elongate and Become Resistant to Mitophagy under Glucose Withdrawal
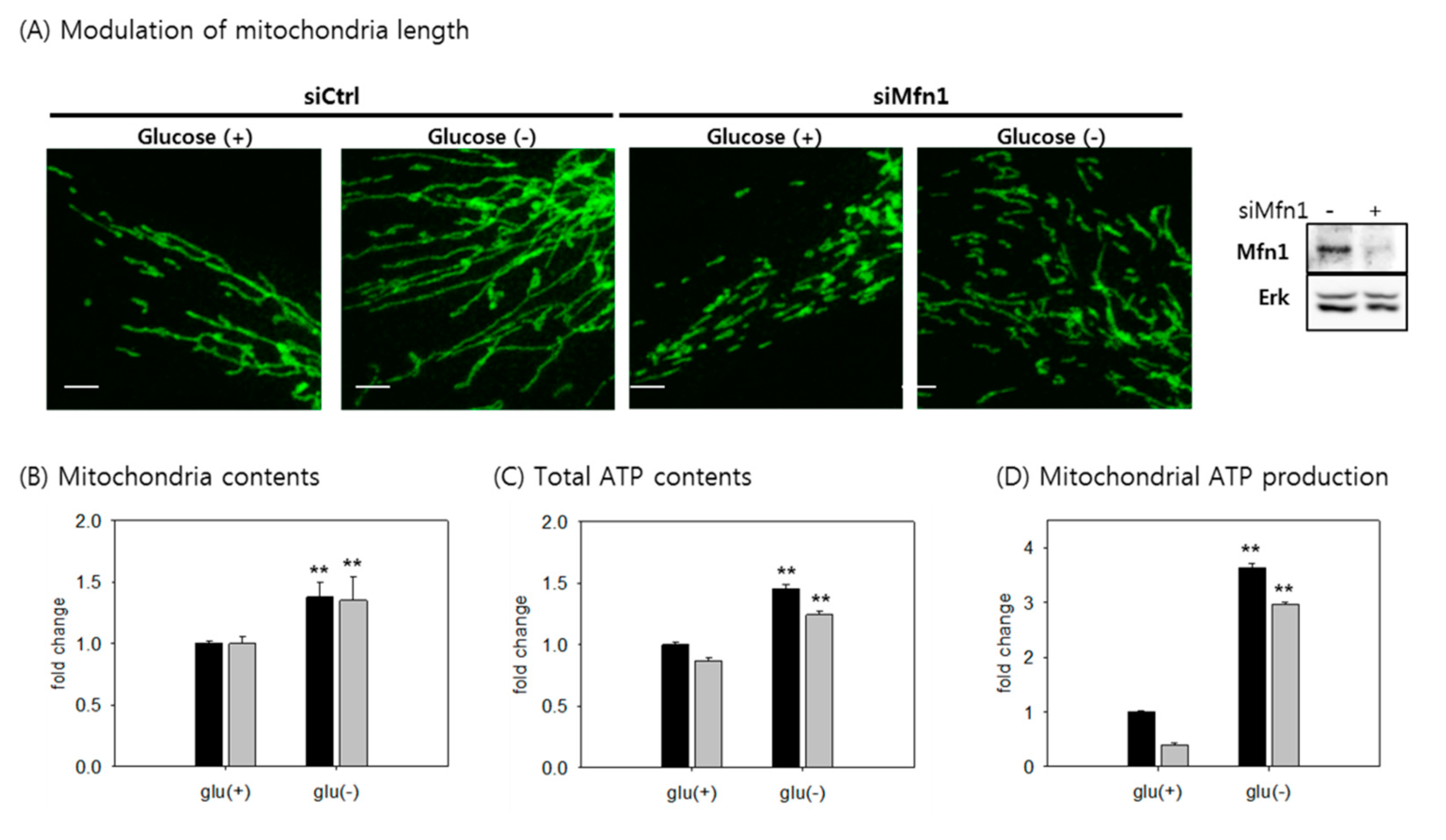
3.7. The Increase in Mitochondrial ATP Synthesis upon Glucose Withdrawal is Attributed to Increase in the Content of Mitochondria
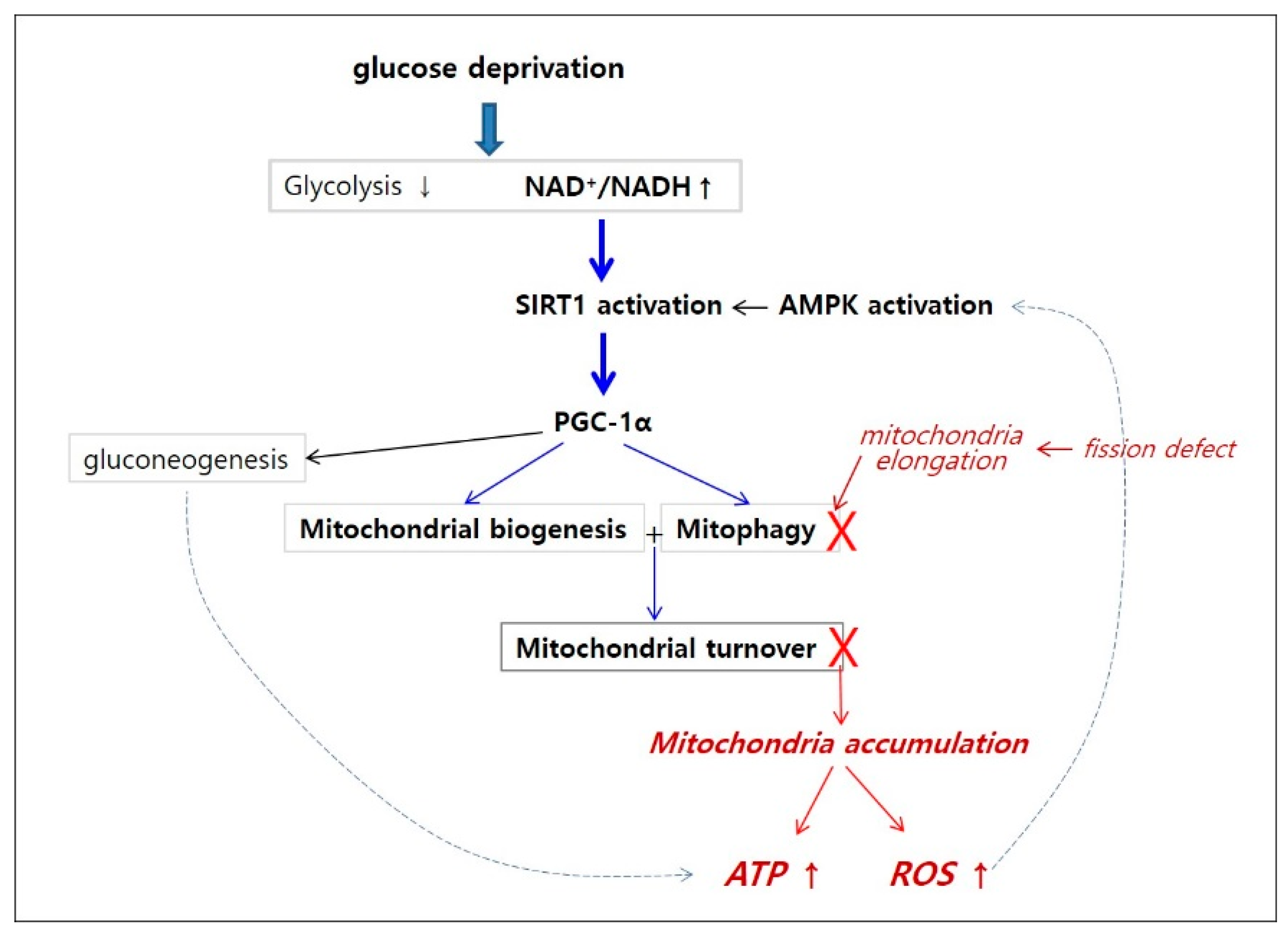
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardie, D.G. Amp-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated protein kinase. Science 2011, 332, 1433–1435. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moley, K.H.; Mueckler, M.M. Glucose transport and apoptosis. Apoptosis 2000, 5, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Demetrakopoulos, G.E.; Linn, B.; Amos, H. Starvation, deoxy-sugars, ouabain, and ATP metabolism in normal and malignant cells. Cancer Biochem. Biophys. 1982, 6, 65–74. [Google Scholar] [PubMed]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [PubMed]
- Buzzai, M.; Bauer, D.E.; Jones, R.G.; Deberardinis, R.J.; Hatzivassiliou, G.; Elstrom, R.L.; Thompson, C.B. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene 2005, 24, 4165–4173. [Google Scholar] [CrossRef]
- Blackburn, R.V.; Spitz, D.R.; Liu, X.; Galoforo, S.S.; Sim, J.E.; Ridnour, L.A.; Chen, J.C.; Davis, B.H.; Corry, P.M.; Lee, Y.J. Metabolic oxidative stress activates signal transduction and gene expression during glucose deprivation in human tumor cells. Free Radic. Biol. Med. 1999, 26, 419–430. [Google Scholar] [CrossRef]
- Lee, Y.J.; Galoforo, S.S.; Berns, C.M.; Chen, J.C.; Davis, B.H.; Sim, J.E.; Corry, P.M.; Spitz, D.R. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J. Biol. Chem. 1998, 273, 5294–5299. [Google Scholar] [CrossRef]
- Ahmad, I.M.; Aykin-Burns, N.; Sim, J.E.; Walsh, S.A.; Higashikubo, R.; Buettner, G.R.; Venkataraman, S.; Mackey, M.A.; Flanagan, S.W.; Oberley, L.W.; et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J. Biol. Chem. 2005, 280, 4254–4263. [Google Scholar] [CrossRef]
- Jelluma, N.; Yang, X.; Stokoe, D.; Evan, G.I.; Dansen, T.B.; Haas-Kogan, D.A. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol. Cancer Res. 2006, 4, 319–330. [Google Scholar] [CrossRef]
- Liu, Y.; Song, X.D.; Liu, W.; Zhang, T.Y.; Zuo, J. Glucose deprivation induces mitochondrial dysfunction and oxidative stress in PC12 cell line. J. Cell. Mol. Med. 2003, 7, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.; Forsberg, L.J.; Viollet, B.; Brenman, J.E. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy 2009, 5, 1155–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marambio, P.; Toro, B.; Sanhueza, C.; Troncoso, R.; Parra, V.; Verdejo, H.; Garcia, L.; Quiroga, C.; Munafo, D.; Diaz-Elizondo, J.; et al. Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim. Biophys. Acta 2010, 1802, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.; Hardie, D.G. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem. J. 1998, 335, 533–539. [Google Scholar] [CrossRef]
- Healy, D.A.; Watson, R.W.; Newsholme, P. Glucose, but not glutamine, protects against spontaneous and anti-fas antibody-induced apoptosis in human neutrophils. Clin. Sci. 2002, 103, 179–189. [Google Scholar] [CrossRef]
- Moore, C.E.; Omikorede, O.; Gomez, E.; Willars, G.B.; Herbert, T.P. PERK activation at low glucose concentration is mediated by SERCA pump inhibition and confers preemptive cytoprotection to pancreatic beta-cells. Mol. Endocrinol. 2011, 25, 315–326. [Google Scholar] [CrossRef]
- Sakurai, T.; Yang, B.; Takata, T.; Yokono, K. Synaptic adaptation to repeated hypoglycemia depends on the utilization of monocarboxylates in guinea pig hippocampal slices. Diabetes 2002, 51, 430–438. [Google Scholar] [CrossRef]
- Hsu, C.P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating sirt1 through AMPK-mediated regulation of nampt. Dev. Cell. 2008, 14, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Gold, A.J.; Yaffe, S.R. Effects of prolonged starvation on cardiac energy metabolism in the rat. J. Nutr. 1978, 108, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Filomeni, G.; Ciriolo, M.R. Redox implications of AMPK-mediated signal transduction beyond energetic clues. J. Cell. Sci. 2012, 125, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals 2009, 2, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Chertov, A.O.; Holzhausen, L.; Kuok, I.T.; Couron, D.; Parker, E.; Linton, J.D.; Sadilek, M.; Sweet, I.R.; Hurley, J.B. Roles of glucose in photoreceptor survival. J. Biol. Chem. 2011, 286, 34700–34711. [Google Scholar] [CrossRef] [PubMed]
- Bordone, L.; Guarente, L. Calorie restriction, sirt1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell. Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef]
- Canto, C.; Auwerx, J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab. 2009, 20, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol. Cell. 2015, 57, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Mitochondria–a nexus for aging, calorie restriction, and sirtuins? Cell 2008, 132, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Jendrach, M.; Pohl, S.; Voth, M.; Kowald, A.; Hammerstein, P.; Bereiter-Hahn, J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.Y.; Kang, H.T.; Hwang, E.S. Nicotinamide-induced mitophagy: Event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 2012, 287, 19304–19314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Marsboom, G.; Toth, P.T.; Rehman, J. Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Tarrado-Castellarnau, M.; de Atauri, P.; Tarrago-Celada, J.; Perarnau, J.; Yuneva, M.; Thomson, T.M.; Cascante, M. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol. Syst Biol 2017, 13. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liang, B.; Shirwany, N.A.; Zou, M.H. 2-deoxy-d-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. Hsir2(sirt1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Chu, C.T. A pivotal role for PINK1 and autophagy in mitochondrial quality control: Implications for Parkinson disease. Hum. Mol. Genet. 2010, 19, R28–R37. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of sirt1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef]
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J. Biol. Chem. 2005, 280, 16456–16460. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell. Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Meley, D.; Bauvy, C.; Houben-Weerts, J.H.; Dubbelhuis, P.F.; Helmond, M.T.; Codogno, P.; Meijer, A.J. AMP-activated protein kinase and the regulation of autophagic proteolysis. J. Biol. Chem. 2006, 281, 34870–34879. [Google Scholar] [CrossRef]
- Law, B.Y.; Wang, M.; Ma, D.L.; Al-Mousa, F.; Michelangeli, F.; Cheng, S.H.; Ng, M.H.; To, K.F.; Mok, A.Y.; Ko, R.Y.; et al. Alisol b, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol. Cancer Ther. 2010, 9, 718–730. [Google Scholar] [CrossRef]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, A.S.; Strack, S. PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics. J. Neurosci. 2011, 31, 15716–15726. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell. Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin e buildup and entry into s phase. Proc. Natl Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef]
- Rolland, S.G.; Motori, E.; Memar, N.; Hench, J.; Frank, S.; Winklhofer, K.F.; Conradt, B. Impaired complex iv activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proc. Natl. Acad. Sci. USA 2013, 110, E2967–E2976. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. Sirtuins: A conserved key unlocking acecs activity. Trends Biochem. Sci. 2007, 32, 1–4. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Borradaile, N.M.; Pickering, J.G. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell. 2009, 8, 100–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourmente, M.; Roldan, E.R. Mass-specific metabolic rate influences sperm performance through energy production in mammals. PLoS ONE 2015, 10, e0138185. [Google Scholar] [CrossRef]
- Pham, A.H.; Chan, D.C. Analyzing mitochondrial dynamics in mouse organotypic slice cultures. Methods Enzymol. 2014, 547, 111–129. [Google Scholar]
- Layec, G.; Bringard, A.; Le Fur, Y.; Micallef, J.P.; Vilmen, C.; Perrey, S.; Cozzone, P.J.; Bendahan, D. Opposite effects of hyperoxia on mitochondrial and contractile efficiency in human quadriceps muscles. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R724–R733. [Google Scholar] [CrossRef] [PubMed]
- Brand, K.A.; Hermfisse, U. Aerobic glycolysis by proliferating cells: A protective strategy against reactive oxygen species. FASEB J. 1997, 11, 388–395. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer. 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kulp, S.K.; Chen, C.S. Energy restriction as an antitumor target of thiazolidinediones. J. Biol. Chem. 2010, 285, 9780–9791. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Scorrano, L. Mitochondrial elongation during autophagy: A stereotypical response to survive in difficult times. Autophagy 2011, 7, 1251–1253. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. Sirt3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.B.; Hwang, E.S. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells 2019, 8, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8010011
Song SB, Hwang ES. A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1. Cells. 2019; 8(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8010011
Chicago/Turabian StyleSong, Seon Beom, and Eun Seong Hwang. 2019. "A Rise in ATP, ROS, and Mitochondrial Content upon Glucose Withdrawal Correlates with a Dysregulated Mitochondria Turnover Mediated by the Activation of the Protein Deacetylase SIRT1" Cells 8, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8010011