Helicobacter pylori Induces IL-33 Production and Recruits ST-2 to Lipid Rafts to Exacerbate Inflammation

 ,
,
Abstract
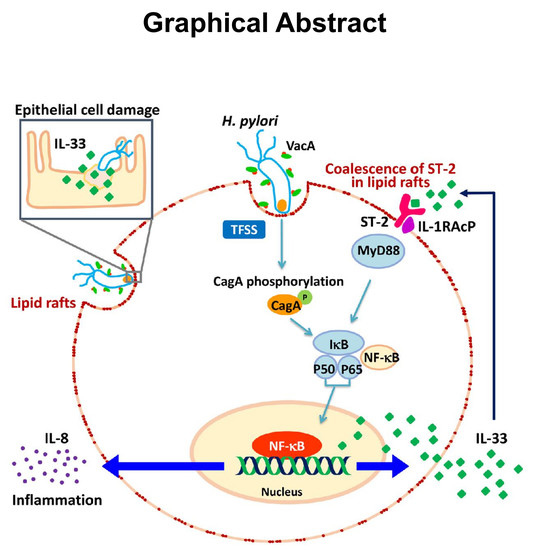
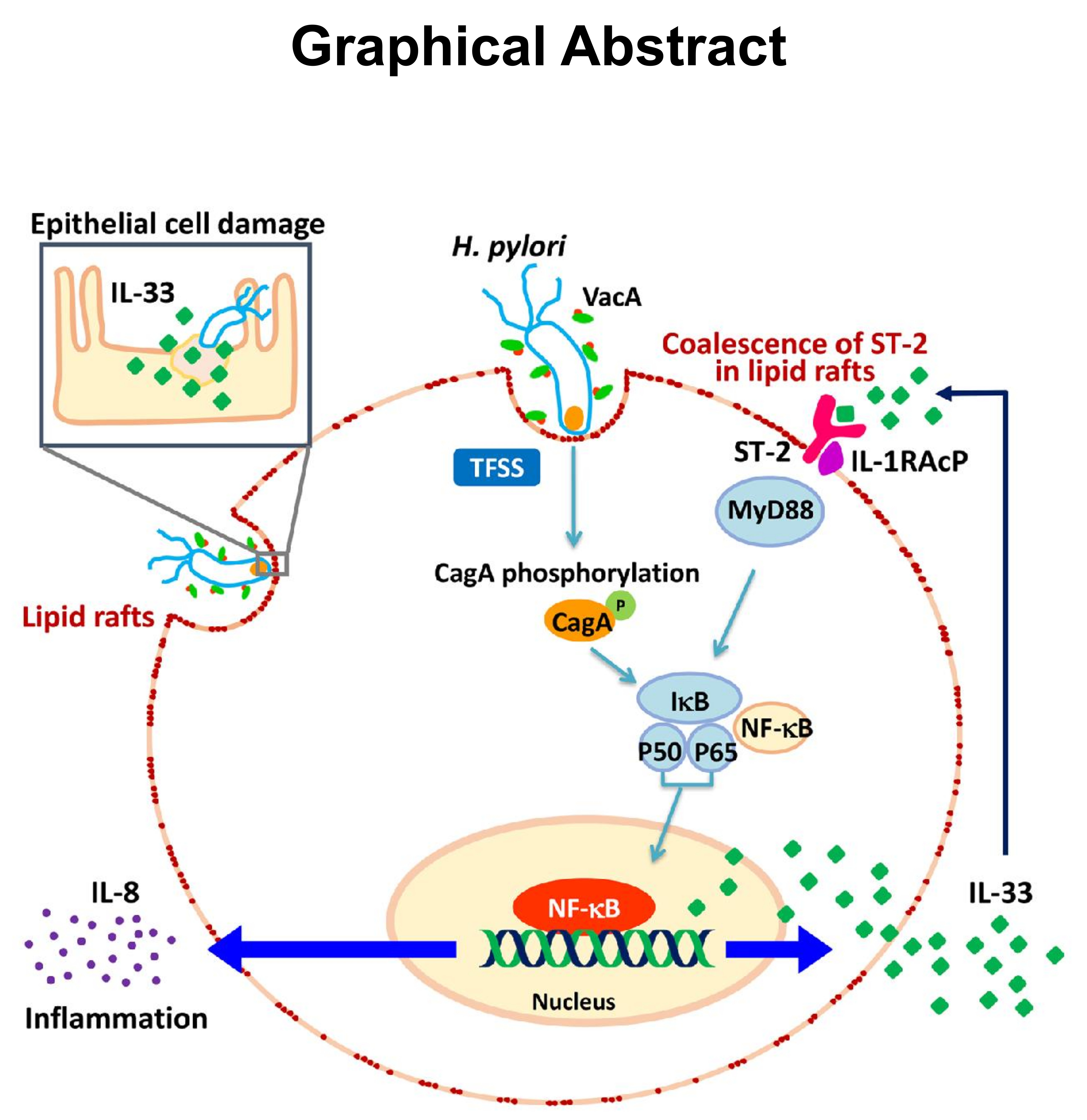
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. H. pylori and Cell Culture
2.2. Western Blot Analysis
2.3. Quantitative Real-Time Reverse Transcription-PCR
2.4. Immunofluorescence Labeling and Confocal Microscopic Analysis
2.5. Fractionation of Cytoplasmic and Nuclear Proteins
2.6. Determination of Cytokine Production
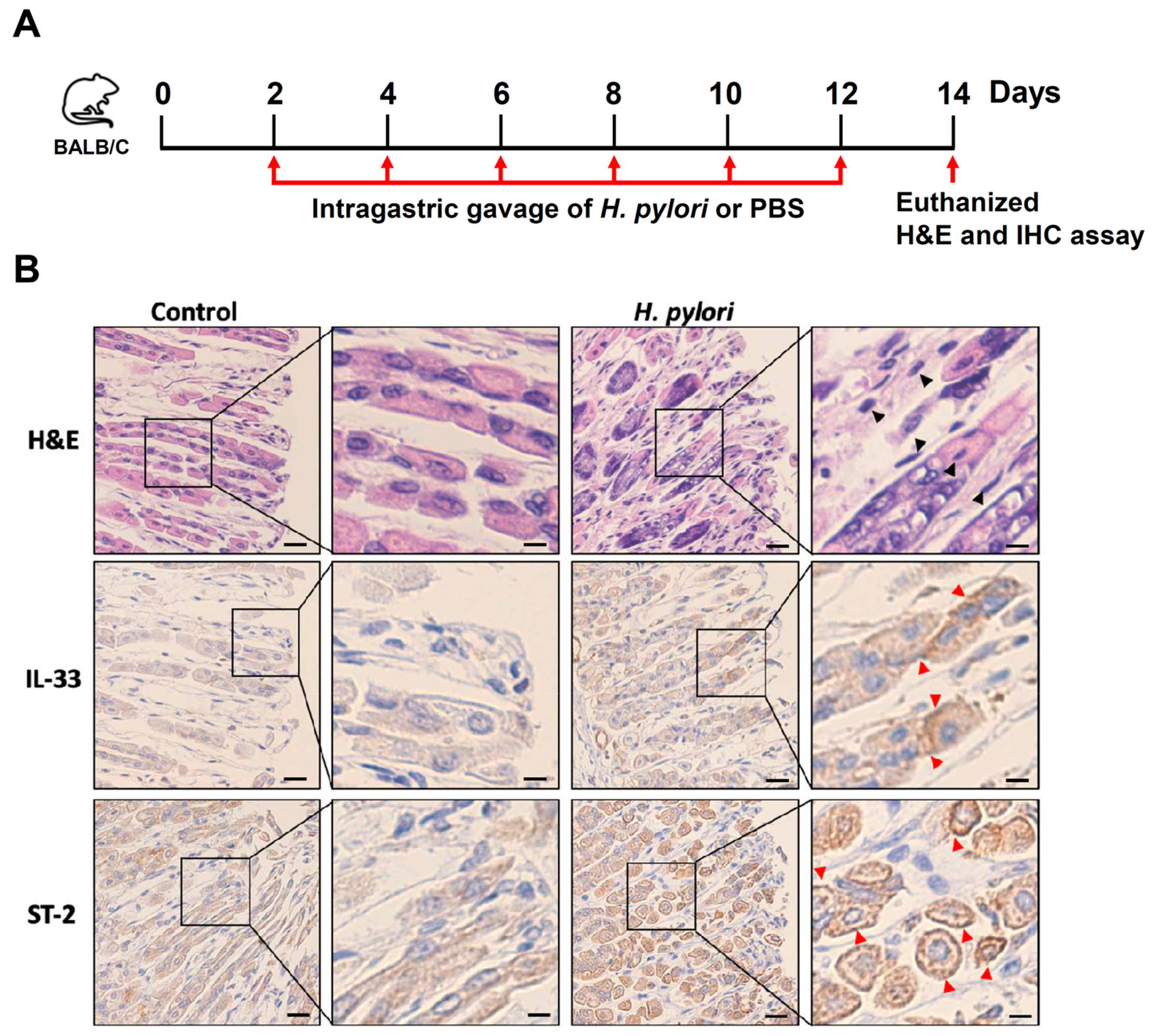
2.7. Animal Study
2.8. Statistical Analysis
3. Results
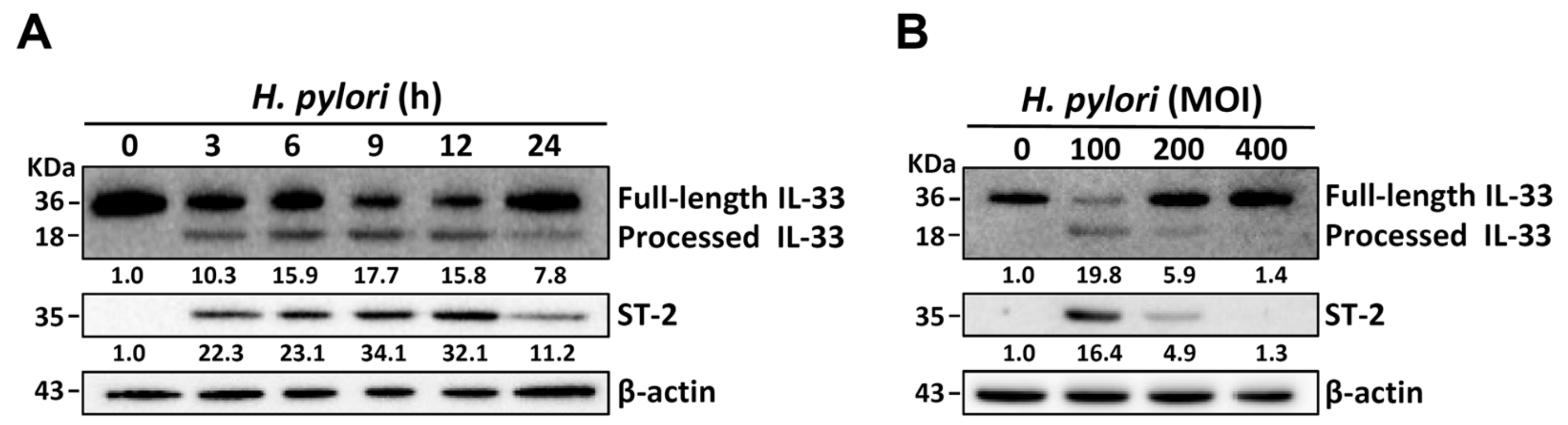
3.1. H. pylori Induces IL-33 Expression in Human Gastric Epithelial Cells
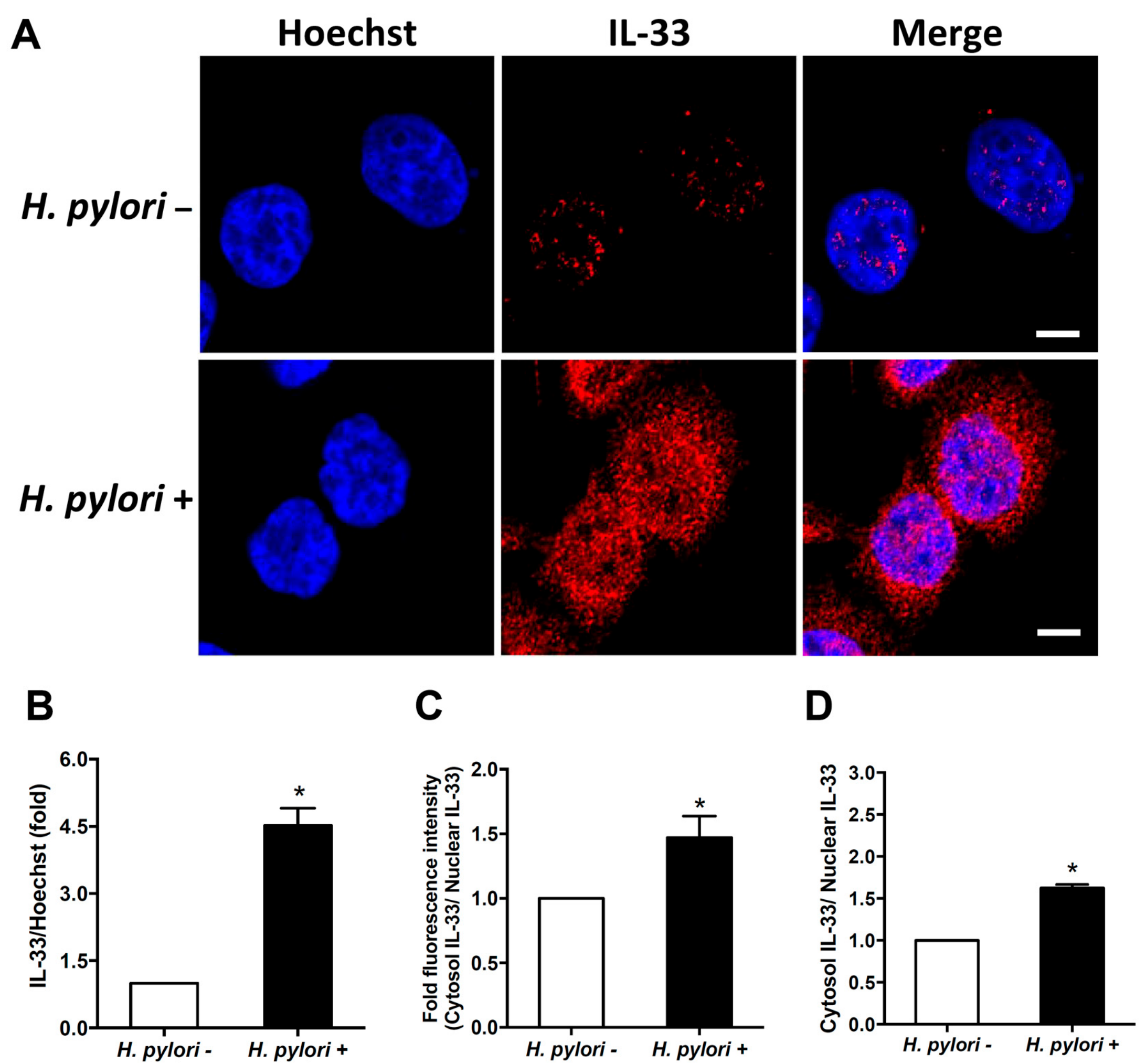
3.2. H. pylori Induces IL-33 Translocation from the Nucleus to the Cytoplasm
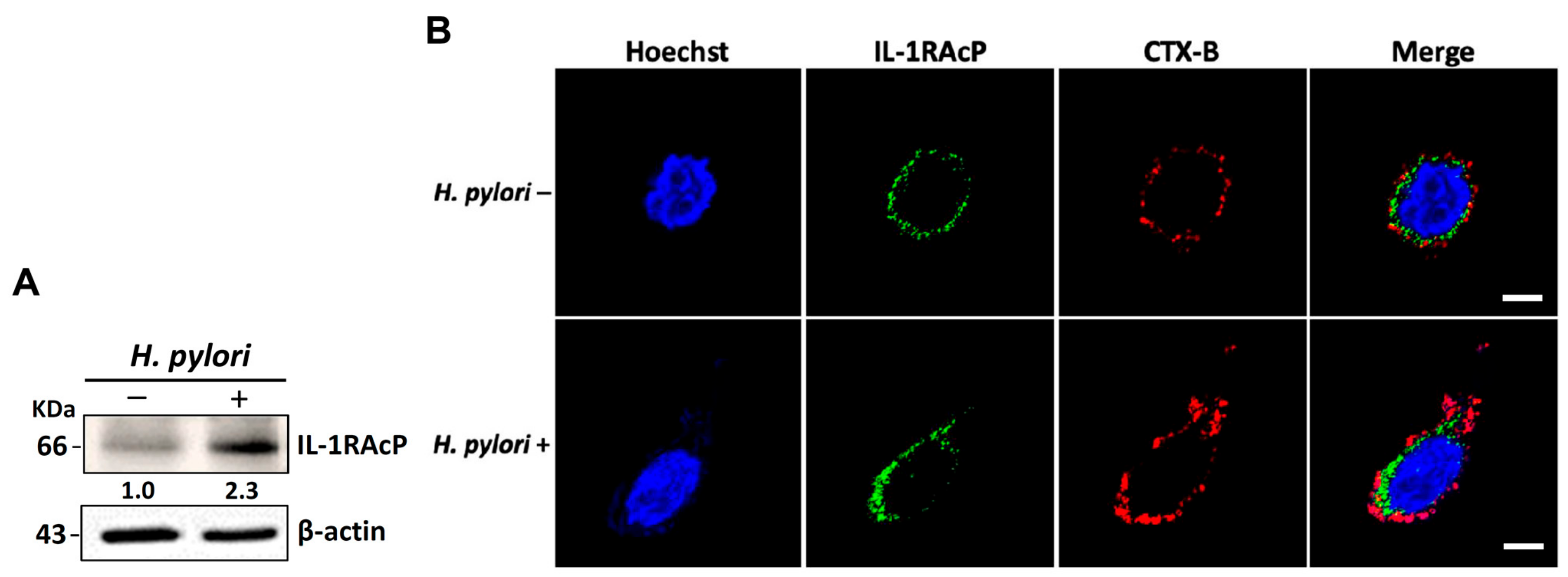
3.3. H. pylori Increases the Levels of IL-33 Receptor ST2 and Co-Receptor IL-1RAcP
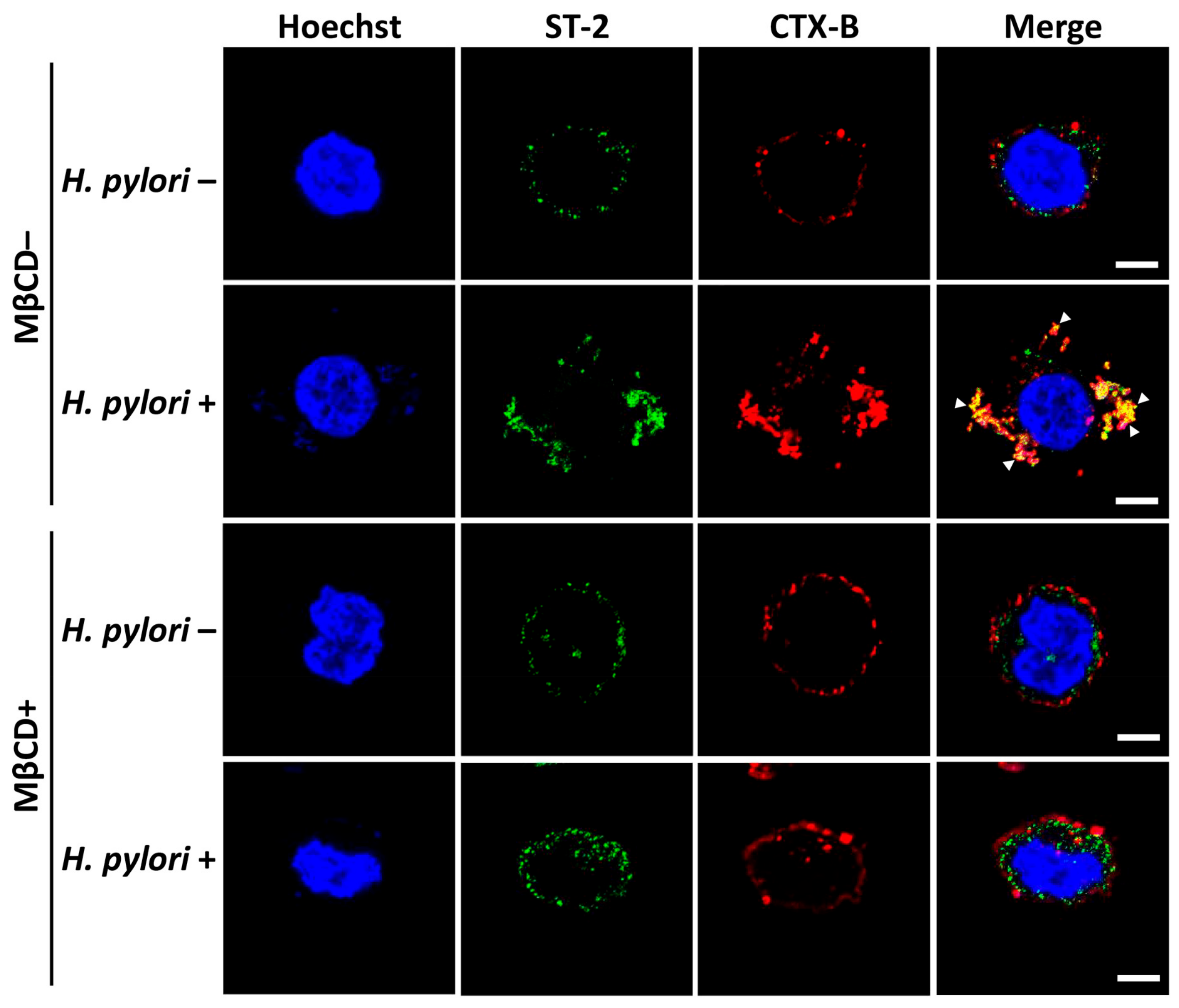
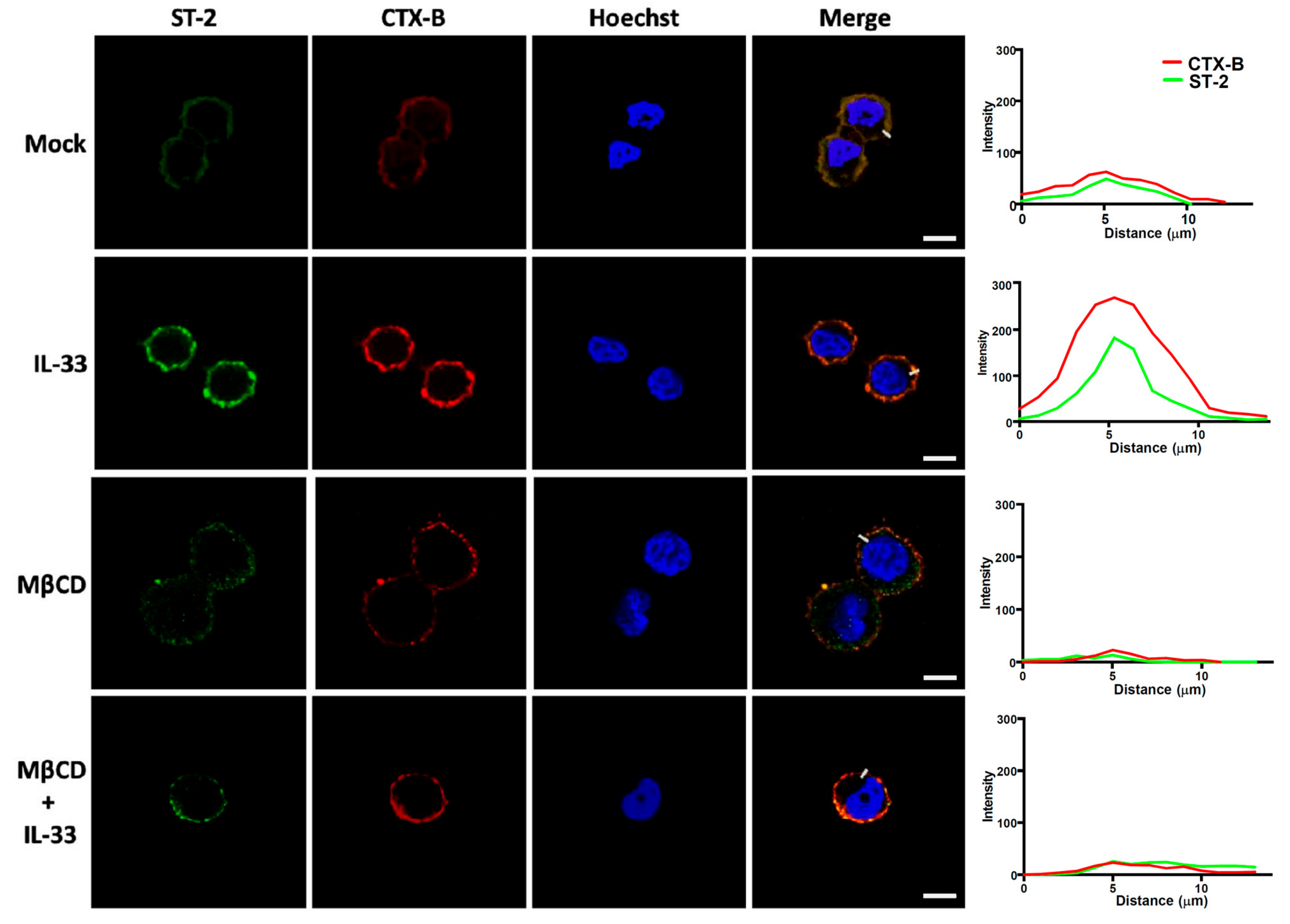
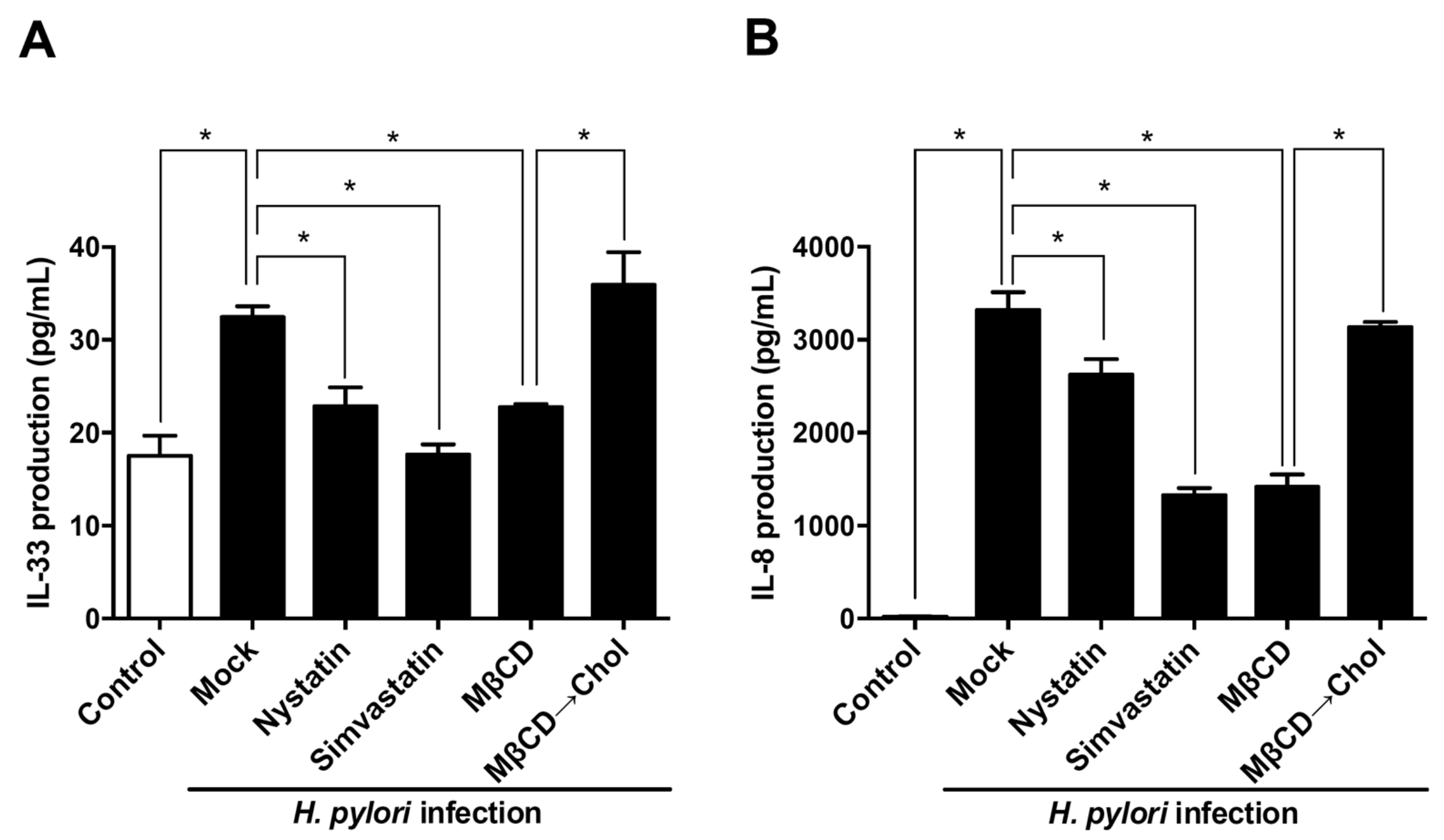
3.4. Sufficient Cholesterol is Crucial for H. pylori-Induced IL-8 and IL-33 Production
3.5. H. pylori Increases the Level of IL-33 Receptor ST2 and Co-Receptor IL-1RAcP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Portal-Celhay, C.; Perez-Perez, G.I. Immune responses to Helicobacter pylori colonization: Mechanisms and clinical outcomes. Clin. Sci. 2006, 110, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Winter, J.A.; Robinson, K. Differential inflammatory response to Helicobacter pylori infection: Etiology and clinical outcomes. J. Inflamm. Res. 2015, 8, 137–147. [Google Scholar] [PubMed]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Pitman, N.I.; McInnes, I.B. Disease-associated functions of IL-33: The new kid in the IL-1 family. Nat. Rev. Immunol. 2010, 10, 103–110. [Google Scholar] [CrossRef]
- Rostan, O.; Arshad, M.I.; Piquet-Pellorce, C.; Robert-Gangneux, F.; Gangneux, J.P.; Samson, M. Crucial and diverse role of the interleukin-33/ST2 axis in infectious diseases. Infect. Immun. 2015, 83, 1738–1748. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Zhu, Q.; Karki, R.; Guy, C.S.; Vogel, P.; Kanneganti, T.D. IL-33 regulates the iga-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J. Clin. Investig. 2016, 126, 4469–4481. [Google Scholar] [CrossRef]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Shahi, H.; Reiisi, S.; Bahreini, R.; Bagheri, N.; Salimzadeh, L.; Shirzad, H. Association between Helicobacter pylori CagA, BabA2 virulence factors and gastric mucosal interleukin-33 mrna expression and clinical outcomes in dyspeptic patients. Int. J. Mol. Cell. Med. 2015, 4, 227–234. [Google Scholar]
- Tran, L.S.; Tran, D.; De Paoli, A.; D’Costa, K.; Creed, S.J.; Ng, G.Z.; Le, L.; Sutton, P.; Silke, J.; Nachbur, U.; et al. Nod1 is required for Helicobacter pylori induction of IL-33 responses in gastric epithelial cells. Cell. Microbiol. 2018, 20, e12826. [Google Scholar] [CrossRef]
- Lv, Y.P.; Teng, Y.S.; Mao, F.Y.; Peng, L.S.; Zhang, J.Y.; Cheng, P.; Liu, Y.G.; Kong, H.; Wang, T.T.; Wu, X.L.; et al. . Helicobacter pylori-induced IL-33 modulates mast cell responses, benefits bacterial growth, and contributes to gastritis. Cell Death Dis. 2018, 9, 457. [Google Scholar] [CrossRef] [PubMed]
- Lafont, F.; van der Goot, F.G. Bacterial invasion via lipid rafts. Cell. Microbiol. 2005, 7, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Manes, S.; del Real, G.; Martinez, A.C. Pathogens: Raft hijackers. Nat. Rev. Immunol. 2003, 3, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Hsu, Y.M.; Wang, H.J.; Wang, W.C. Manipulation of host cholesterol by Helicobacter pylori for their beneficial ecological niche. BioMedicine 2013, 3, 27–33. [Google Scholar] [CrossRef]
- Lai, C.H.; Chang, Y.C.; Du, S.Y.; Wang, H.J.; Kuo, C.H.; Fang, S.H.; Fu, H.W.; Lin, H.H.; Chiang, A.S.; Wang, W.C. Cholesterol depletion reduces Helicobacter pylori CagA translocation and CagA-induced responses in ags cells. Infect. Immun. 2008, 76, 3293–3303. [Google Scholar] [CrossRef]
- Wunder, C.; Churin, Y.; Winau, F.; Warnecke, D.; Vieth, M.; Lindner, B.; Zahringer, U.; Mollenkopf, H.J.; Heinz, E.; Meyer, T.F. Cholesterol glucosylation promotes immune evasion by Helicobacter pylori. Nat. Med. 2006, 12, 1030–1038. [Google Scholar] [CrossRef]
- Basu, S.; Pathak, S.K.; Chatterjee, G.; Pathak, S.; Basu, J.; Kundu, M. Helicobacter pylori protein hp0175 transactivates epidermal growth factor receptor through tlr4 in gastric epithelial cells. J. Biol. Chem. 2008, 283, 32369–32376. [Google Scholar] [CrossRef]
- Lai, C.H.; Huang, J.C.; Cheng, H.H.; Wu, M.C.; Huang, M.Z.; Hsu, H.Y.; Chen, Y.A.; Hsu, C.Y.; Pan, Y.J.; Chu, Y.T.; et al. Helicobacter pylori cholesterol glucosylation modulates autophagy for increasing intracellular survival in macrophages. Cell. Microbiol. 2018, 20, e12947. [Google Scholar] [CrossRef]
- Lin, C.J.; Liao, W.C.; Lin, H.J.; Hsu, Y.M.; Lin, C.L.; Chen, Y.A.; Feng, C.L.; Chen, C.J.; Kao, M.C.; Lai, C.H.; et al. Statins attenuate Helicobacter pylori CagA translocation and reduce incidence of gastric cancer: In vitro and population-based case-control studies. PLoS ONE 2016, 11, e0146432. [Google Scholar] [CrossRef]
- Lin, H.J.; Hsu, F.Y.; Chen, W.W.; Lee, C.H.; Lin, Y.J.; Chen, Y.Y.; Chen, C.J.; Huang, M.Z.; Kao, M.C.; Chen, Y.A.; et al. Helicobacter pylori activates HMGB11 expression and recruits rage into lipid rafts to promote inflammation in gastric epithelial cells. Front. Immunol. 2016, 7, 341. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Liao, W.C.; Chen, Y.A.; Lin, H.J.; Feng, C.L.; Lin, C.L.; Lin, Y.J.; Kao, M.C.; Huang, M.Z.; Lai, C.H.; et al. Statin therapy is associated with reduced risk of peptic ulcer disease in the taiwanese population. Front. Pharmacol. 2017, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Tomb, J.F.; White, O.; Kerlavage, A.R.; Clayton, R.A.; Sutton, G.G.; Fleischmann, R.D.; Ketchum, K.A.; Klenk, H.P.; Gill, S.; Dougherty, B.A.; et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 1997, 388, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.J.; Jiang, Z.P.; Lo, H.R.; Feng, C.L.; Chen, C.J.; Yang, C.Y.; Huang, M.Z.; Wu, H.Y.; Chen, Y.A.; Chen, Y.; et al. Coalescence of RAGE in lipid rafts in response to cytolethal distending toxin-induced inflammation. Front. Immunol. 2019, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.L.; Gilchrist, D.S.; Akbar, M.; Reilly, J.H.; Kerr, S.C.; Campbell, A.L.; Murrell, G.A.C.; Liew, F.Y.; Kurowska-Stolarska, M.; McInnes, I.B. MicroRNA29a regulates IL-33-mediated tissue remodelling in tendon disease. Nat. Commun. 2015, 6, 6774. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.C.; Huang, M.Z.; Wang, M.L.; Lin, C.J.; Lu, T.L.; Lo, H.R.; Pan, Y.J.; Sun, Y.C.; Kao, M.C.; Lim, H.J.; et al. Statin decreases Helicobacter pylori burden in macrophages by promoting autophagy. Front. Cell. Infect. Microbiol. 2017, 6, 203. [Google Scholar] [CrossRef]
- Lu, D.Y.; Chen, H.C.; Yang, M.S.; Hsu, Y.M.; Lin, H.J.; Tang, C.H.; Lee, C.H.; Lai, C.K.; Lin, C.J.; Shyu, W.C.; et al. Ceramide and toll-like receptor 4 are mobilized into membrane rafts in response to Helicobacter pylori infection in gastric epithelial cells. Infect. Immun. 2012, 80, 1823–1833. [Google Scholar] [CrossRef]
- Huang, J.; Lo, U.G.; Wu, S.; Wang, B.; Pong, R.C.; Lai, C.H.; Lin, H.; He, D.; Hsieh, J.T.; Wu, K. The roles and mechanism of IFIT5 in bladder cancer epithelial-mesenchymal transition and progression. Cell Death Dis. 2019, 10, 437. [Google Scholar] [CrossRef]
- Chen, Y.H.; Tsai, W.H.; Wu, H.Y.; Chen, C.Y.; Yeh, W.L.; Chen, Y.H.; Hsu, H.Y.; Chen, W.W.; Chen, Y.W.; Chang, W.W.; et al. Probiotic Lactobacillus spp. act against Helicobacter pylori-induced inflammation. J. Clin. Med. 2019, 8, 90. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces t helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Hoogerwerf, J.J.; Leendertse, M.; Wieland, C.W.; de Vos, A.F.; de Boer, J.D.; Florquin, S.; van der Poll, T. Loss of suppression of tumorigenicity 2 (ST2) gene reverses sepsis-induced inhibition of lung host defense in mice. Am. J. Respir. Crit. Care Med. 2011, 183, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Buckley, J.M.; Redmond, H.P.; Wang, J.H. St2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J. Immunol. 2010, 184, 5802–5808. [Google Scholar] [CrossRef] [PubMed]
- Farias, R.; Rousseau, S. The Tak1-->Ikkbeta-->Tpl2-->Mkk1/Mkk2 signaling cascade regulates IL-33 expression in cystic fibrosis airway epithelial cells following infection by Pseudomonas aeruginosa. Front. Cell Dev. Biol. 2015, 3, 87. [Google Scholar] [PubMed]
- Li, C.; Li, H.; Jiang, Z.; Zhang, T.; Wang, Y.; Li, Z.; Wu, Y.; Ji, S.; Xiao, S.; Ryffel, B.; et al. Interleukin-33 increases antibacterial defense by activation of inducible nitric oxide synthase in skin. PLoS Pathog. 2014, 10, e1003918. [Google Scholar] [CrossRef] [PubMed]
- Heyen, L.; Muller, U.; Siegemund, S.; Schulze, B.; Protschka, M.; Alber, G.; Piehler, D. Lung epithelium is the major source of IL-33 and is regulated by IL-33-dependent and IL-33-independent mechanisms in pulmonary Cryptococcosis. Pathog. Dis. 2016, 74, 1–11. [Google Scholar] [CrossRef]
- Wagenaar, J.F.; Gasem, M.H.; Goris, M.G.; Leeflang, M.; Hartskeerl, R.A.; van der Poll, T.; van ’t Veer, C.; van Gorp, E.C. Soluble ST2 levels are associated with bleeding in patients with severe leptospirosis. PLoS Negl. Trop. Dis. 2009, 3, e453. [Google Scholar] [CrossRef]
- Yin, H.; Li, X.; Hu, S.; Liu, T.; Yuan, B.; Ni, Q.; Lan, F.; Luo, X.; Gu, H.; Zheng, F. IL-33 promotes Staphylococcus aureus-infected wound healing in mice. Int. Immunopharmacol. 2013, 17, 432–438. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; Sonego, F.; Souto, F.O.; Freitas, A.; Verri, W.A., Jr.; Auxiliadora-Martins, M.; Basile-Filho, A.; McKenzie, A.N.; Xu, D.; Cunha, F.Q.; et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 2010, 16, 708–712. [Google Scholar] [CrossRef]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef]
- Moulin, D.; Donze, O.; Talabot-Ayer, D.; Mezin, F.; Palmer, G.; Gabay, C. Interleukin (IL)-33 induces the release of pro-inflammatory mediators by mast cells. Cytokine 2007, 40, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Taracanova, A.; Alevizos, M.; Karagkouni, A.; Weng, Z.; Norwitz, E.; Conti, P.; Leeman, S.E.; Theoharides, T.C. Sp and IL-33 together markedly enhance tnf synthesis and secretion from human mast cells mediated by the interaction of their receptors. Proc. Natl. Acad. Sci. USA 2017, 114, E4002–E4009. [Google Scholar] [CrossRef] [PubMed]
- Yakabi, K.; Arimura, T.; Koyanagi, M.; Uehigashi, Y.; Ro, S.; Minagawa, Y.; Nakamura, T. Effects of interleukin-8 and Helicobacter pylori on histamine release from isolated canine gastric mucosal mast cells. J. Gastroenterol. 2002, 37, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, D.C.; Melo, P.H.; Pineros, A.R.; Ferreira, R.G.; Colon, D.F.; Donate, P.B.; Castanheira, F.V.; Gozzi, A.; Czaikoski, P.G.; Niedbala, W.; et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory t cell population. Nat. Commun. 2017, 8, 14919. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Buzzelli, J.N.; Chalinor, H.V.; Pavlic, D.I.; Sutton, P.; Menheniott, T.R.; Giraud, A.S.; Judd, L.M. IL33 is a stomach alarmin that initiates a skewed Th2 response to injury and infection. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 203–221. [Google Scholar] [CrossRef]
- Noto, J.M.; Peek, R.M., Jr. Sound the alarmin: Interleukin-33 commandeers the gastric immune response. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 127–128. [Google Scholar] [CrossRef]
- Motzkus-Feagans, C.A.; Pakyz, A.; Polk, R.; Gambassi, G.; Lapane, K.L. Statin use and the risk of clostridium difficile in academic medical centres. Gut 2012, 61, 1538–1542. [Google Scholar] [CrossRef]
- Boyd, A.R.; Hinojosa, C.A.; Rodriguez, P.J.; Orihuela, C.J. Impact of oral simvastatin therapy on acute lung injury in mice during pneumococcal pneumonia. BMC Microbiol. 2012, 12, 73. [Google Scholar] [CrossRef]
- Chow, O.A.; von Kockritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef]
- Parihar, S.P.; Guler, R.; Khutlang, R.; Lang, D.M.; Hurdayal, R.; Mhlanga, M.M.; Suzuki, H.; Marais, A.D.; Brombacher, F. Statin therapy reduces the mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 2014, 209, 754–763. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, L.; Hua, S.; Qi, C.; Fang, M. IL-33 ameliorates experimental colitis involving regulation of autophagy of macrophages in mice. Cell Biosci. 2019, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.D.; Alevy, Y.; Malvin, N.P.; Patel, K.K.; Gunsten, S.P.; Holtzman, M.J.; Stappenbeck, T.S.; Brody, S.L. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy 2016, 12, 397–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Huang, M.; Yao, Y.M. Autophagy and proinflammatory cytokines: Interactions and clinical implications. Cytokine Growth Factor Rev. 2018, 43, 38–46. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, C.-J.; Chen, C.-Y.; Lo, H.-R.; Feng, C.-L.; Wu, H.-Y.; Huang, M.-Z.; Liao, T.-N.; Chen, Y.-A.; Lai, C.-H. Helicobacter pylori Induces IL-33 Production and Recruits ST-2 to Lipid Rafts to Exacerbate Inflammation. Cells 2019, 8, 1290. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101290
Kuo C-J, Chen C-Y, Lo H-R, Feng C-L, Wu H-Y, Huang M-Z, Liao T-N, Chen Y-A, Lai C-H. Helicobacter pylori Induces IL-33 Production and Recruits ST-2 to Lipid Rafts to Exacerbate Inflammation. Cells. 2019; 8(10):1290. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101290
Chicago/Turabian StyleKuo, Chia-Jung, Chun-Ya Chen, Horng-Ren Lo, Chun-Lung Feng, Hui-Yu Wu, Mei-Zi Huang, Tung-Nan Liao, Yu-An Chen, and Chih-Ho Lai. 2019. "Helicobacter pylori Induces IL-33 Production and Recruits ST-2 to Lipid Rafts to Exacerbate Inflammation" Cells 8, no. 10: 1290. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8101290