Exogenous Cytokine-Free Differentiation of Human Pluripotent Stem Cells into Classical Brown Adipocytes


Abstract
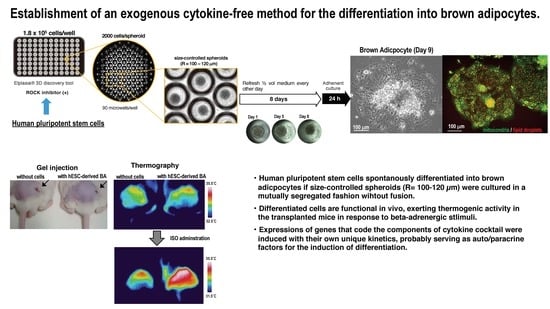
:
1. Introduction
2. Materials and Methods
2.1. Immature Maintenance of Human Pluripotent Stem Cells
2.2. Differentiation into Classical BA
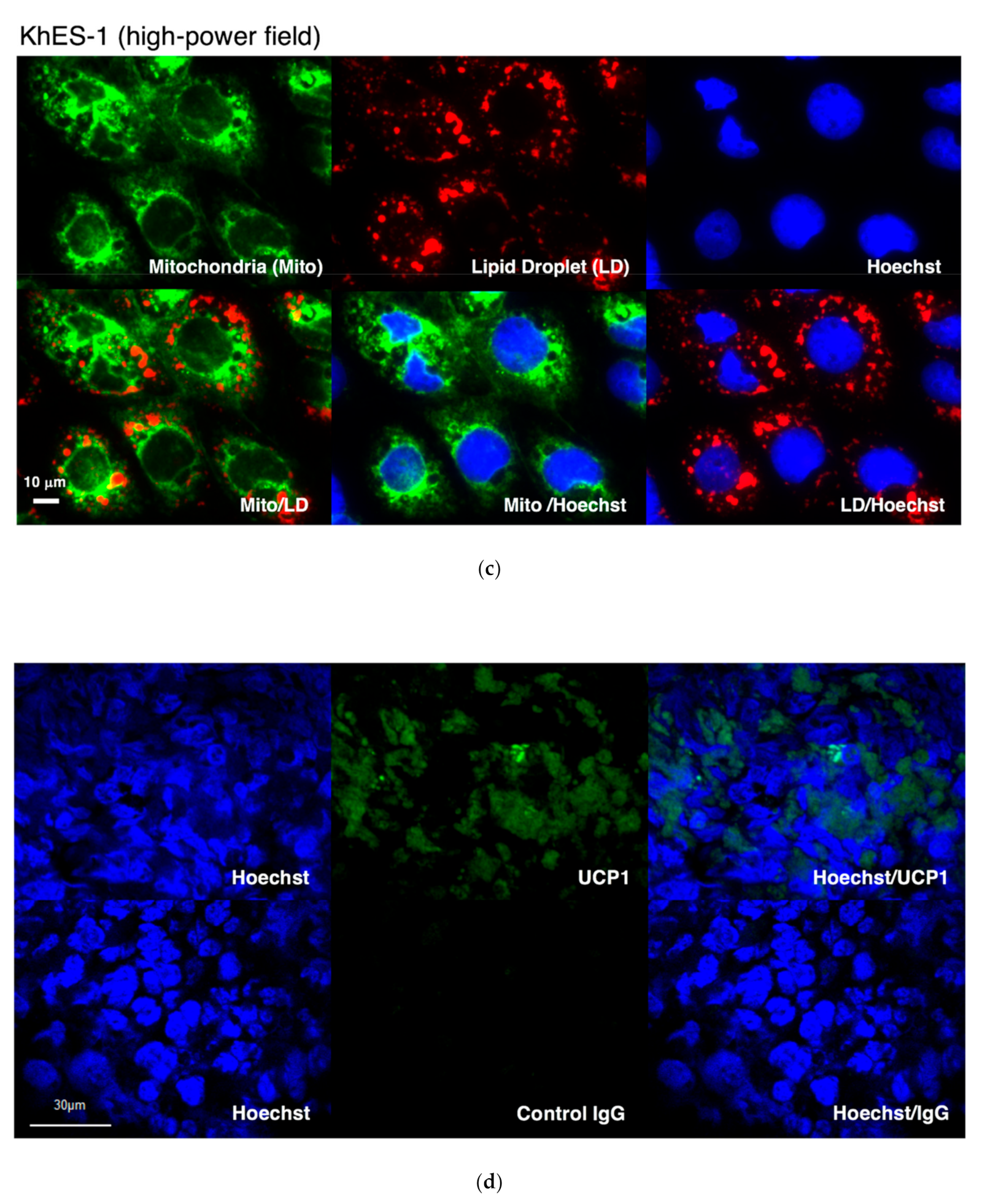
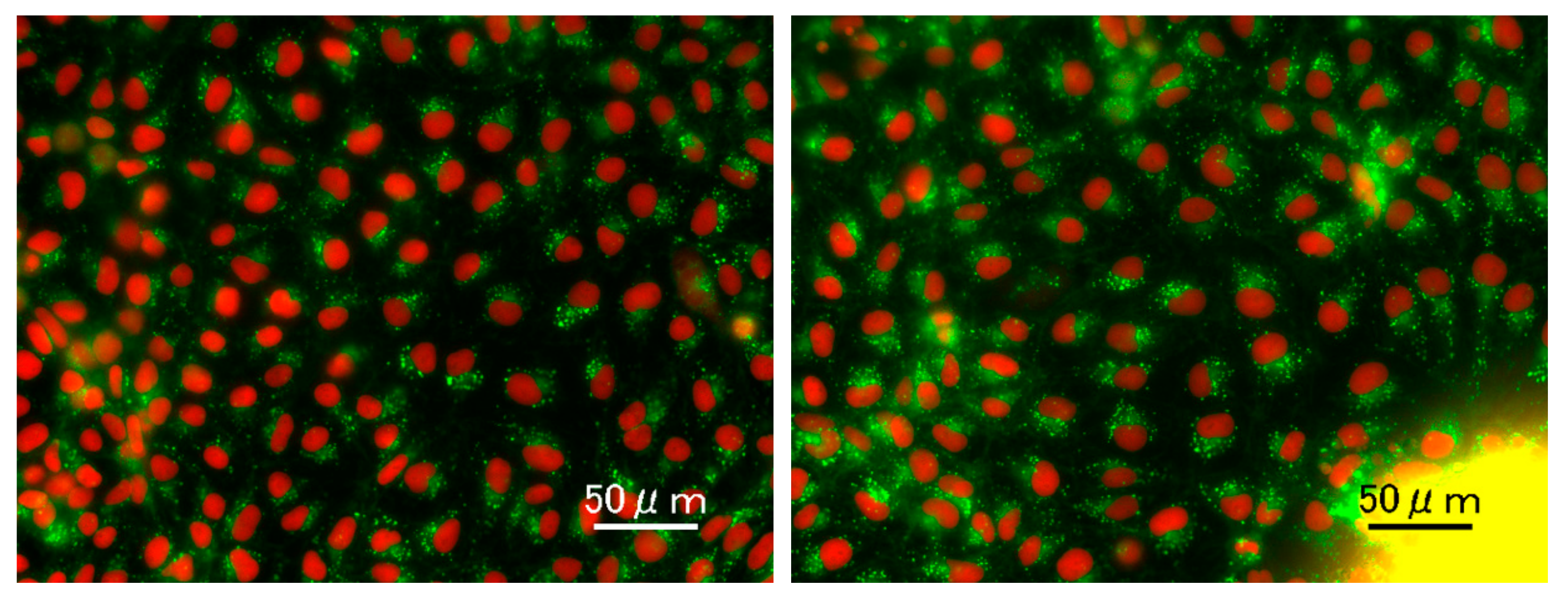
2.3. Live Cell Staining of Mitochondria and Lipid Droplets
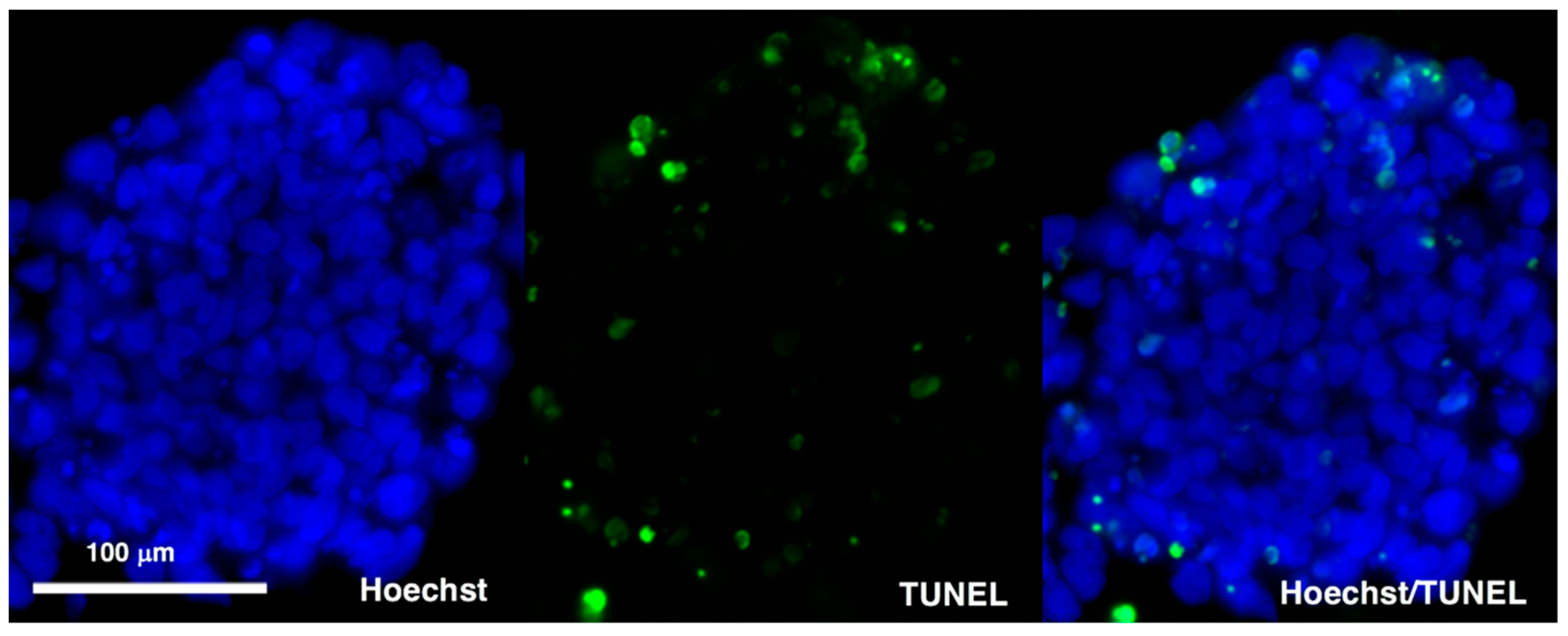
2.4. Immunostaining and TUNEL Assays
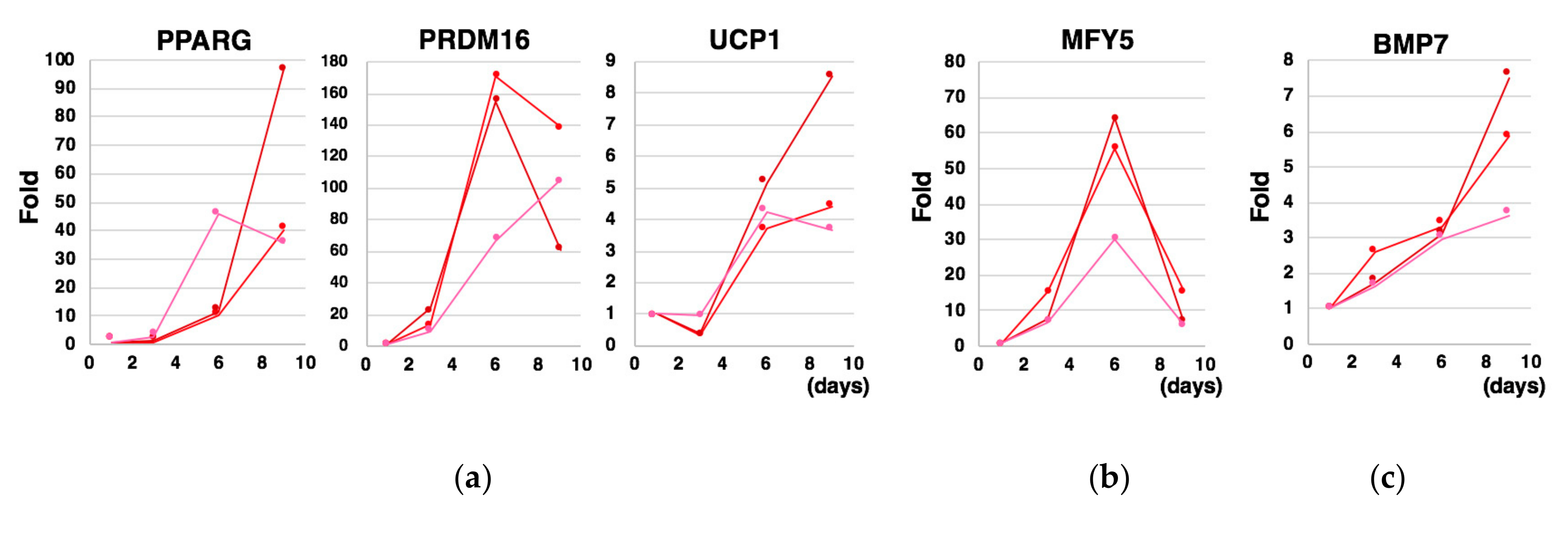
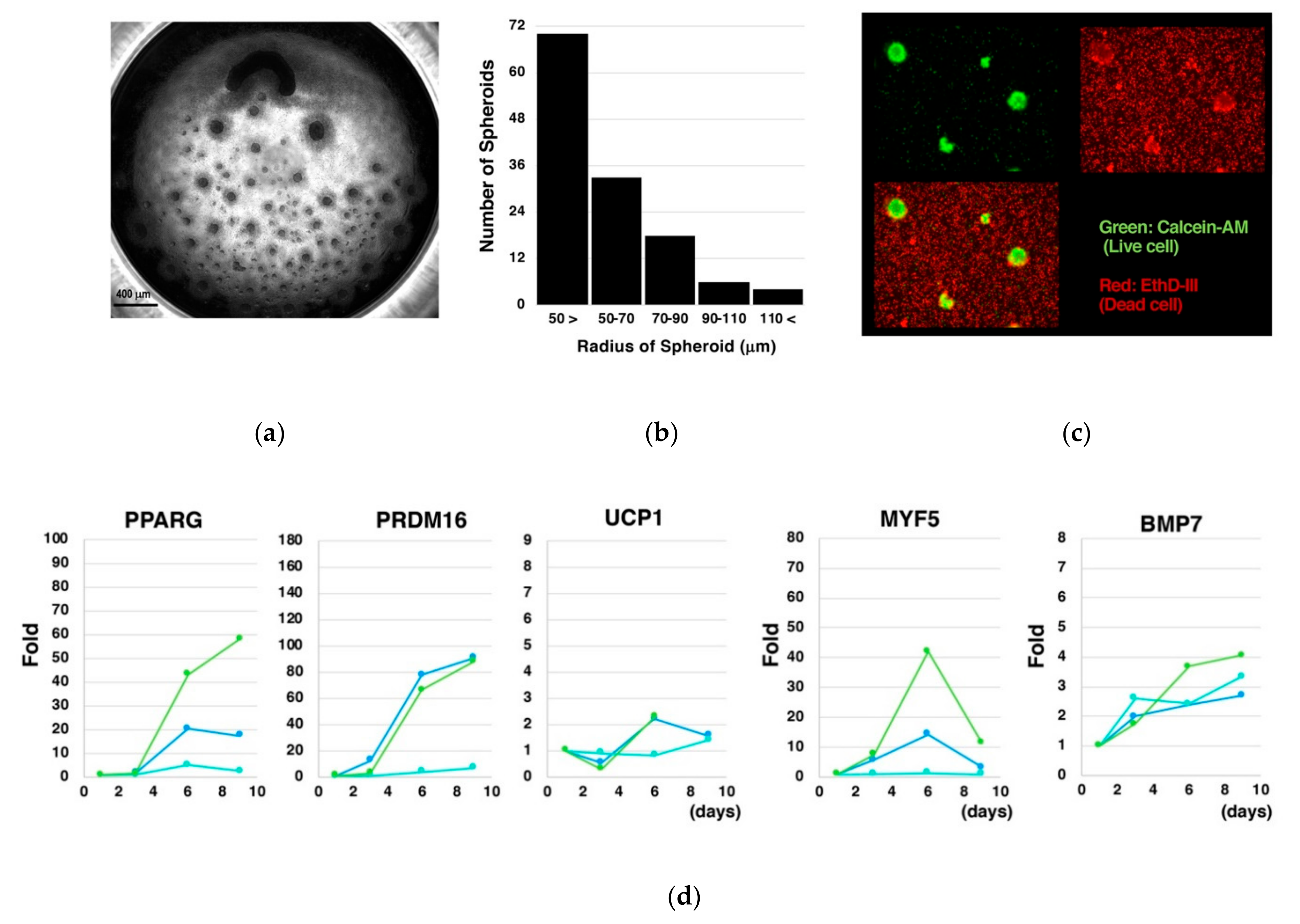
2.5. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.6. In Vivo Calorigenic Analyses
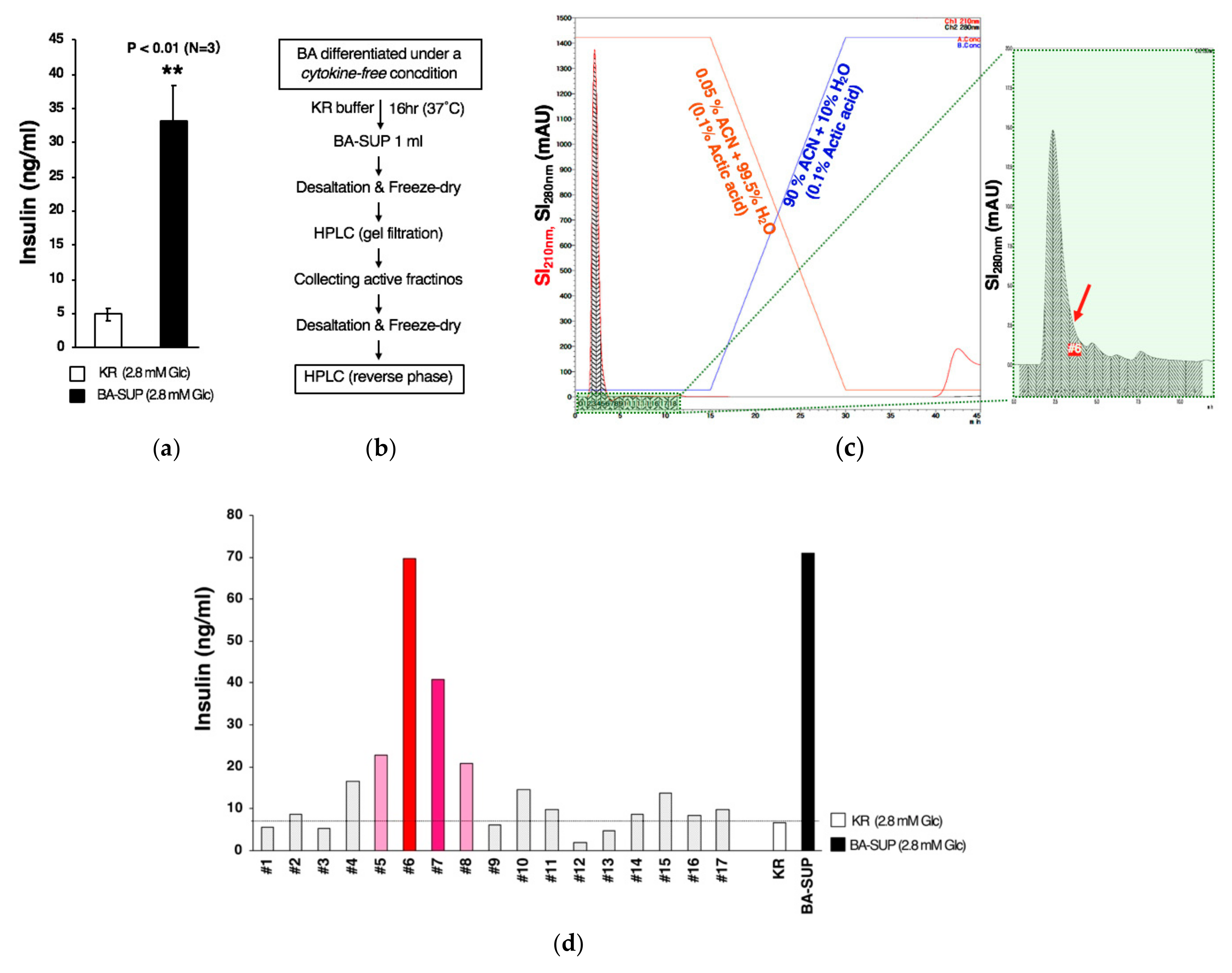
2.7. Preparation of the Supernatant of BA (BA-SUP) and an Evaluation of its Biological Activity
2.8. Insulin Secretion Assay In Vitro
2.9. Statistics
2.10. Partial Purification of a Novel Insulin Secretion-Stimulating Factor from BA-SUP by Using High-Pressure Liquid Chromatography (HPLC)
2.11. Establishment of Knock-In hESC Line
3. Results
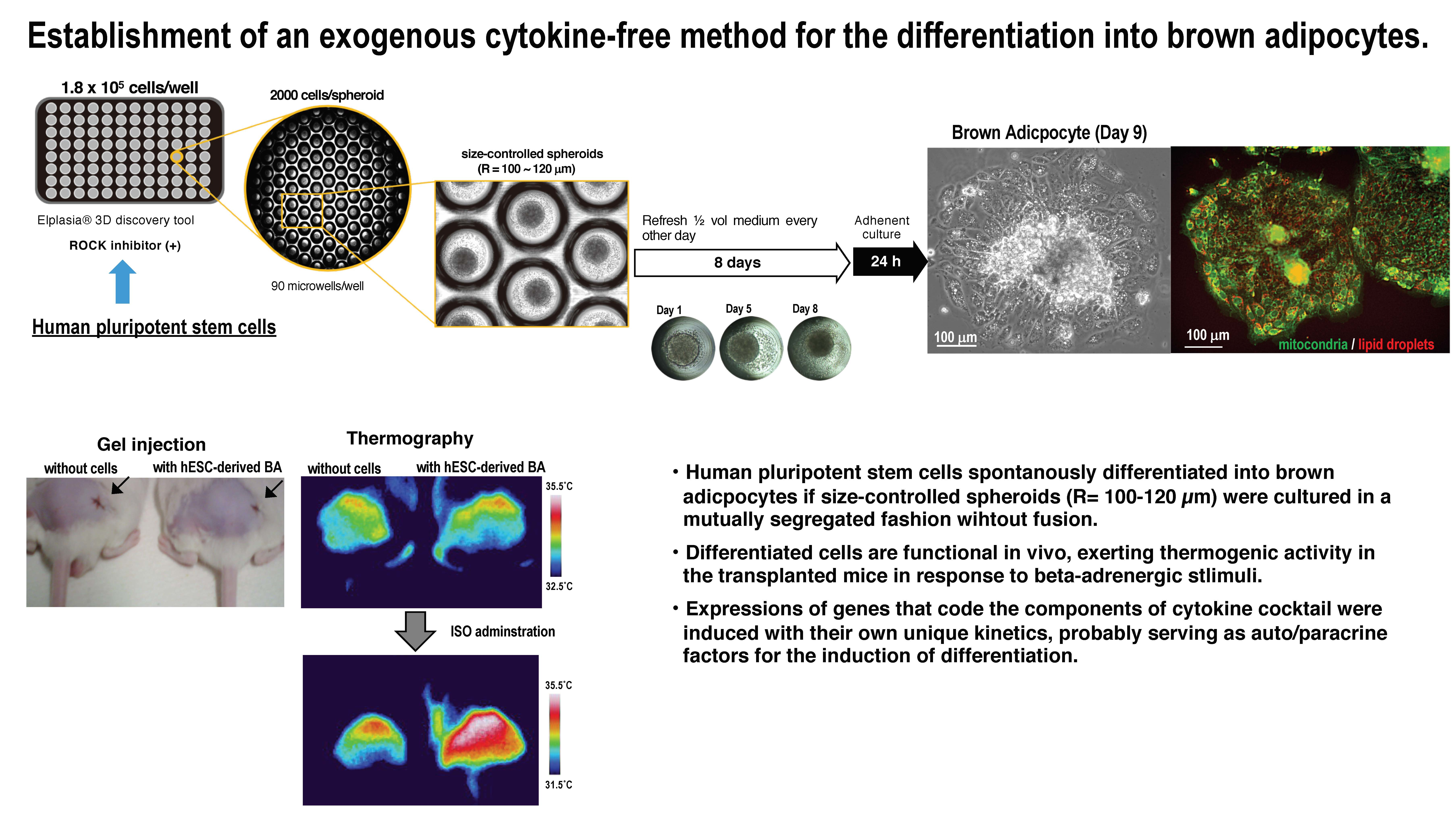
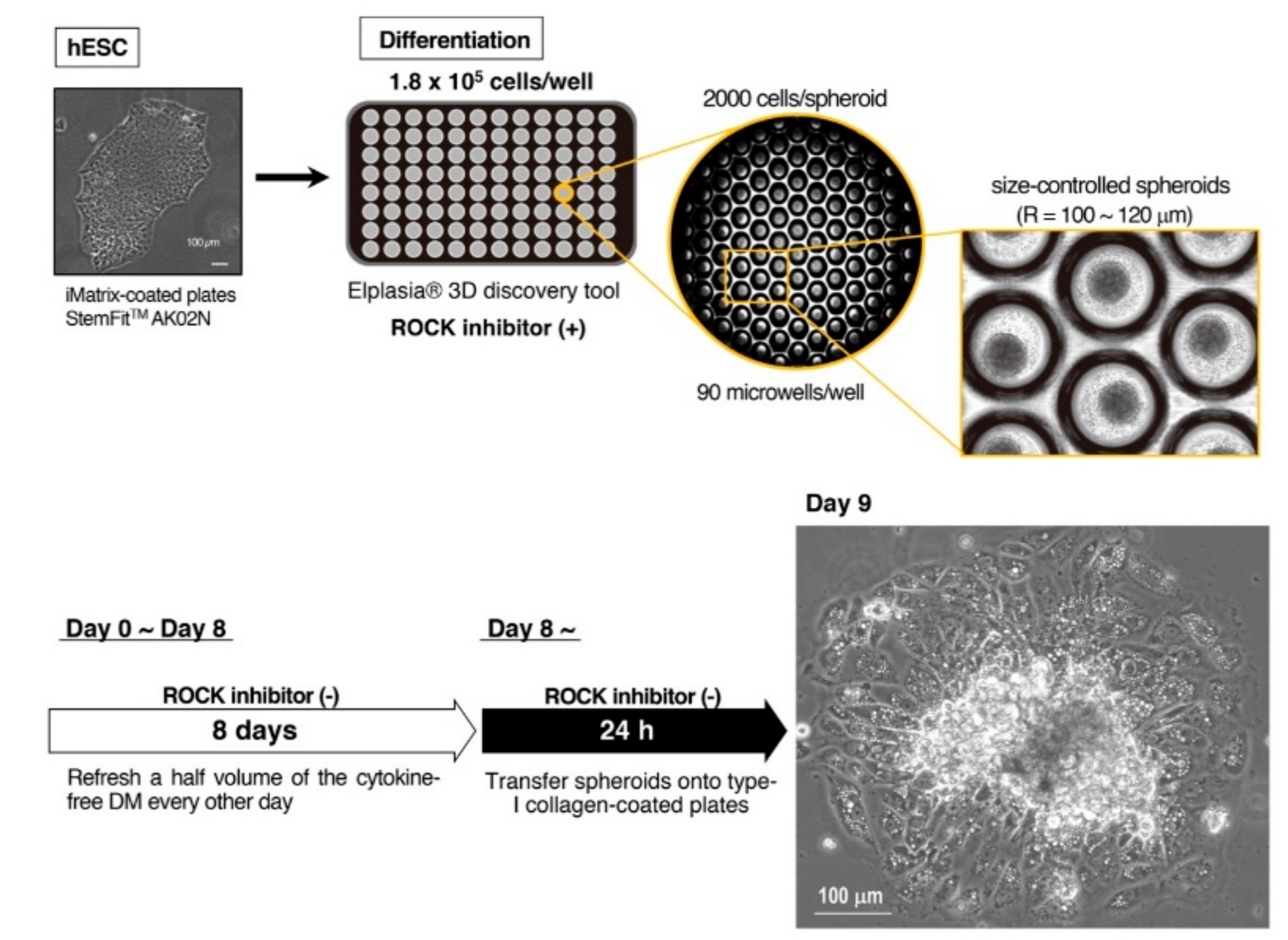
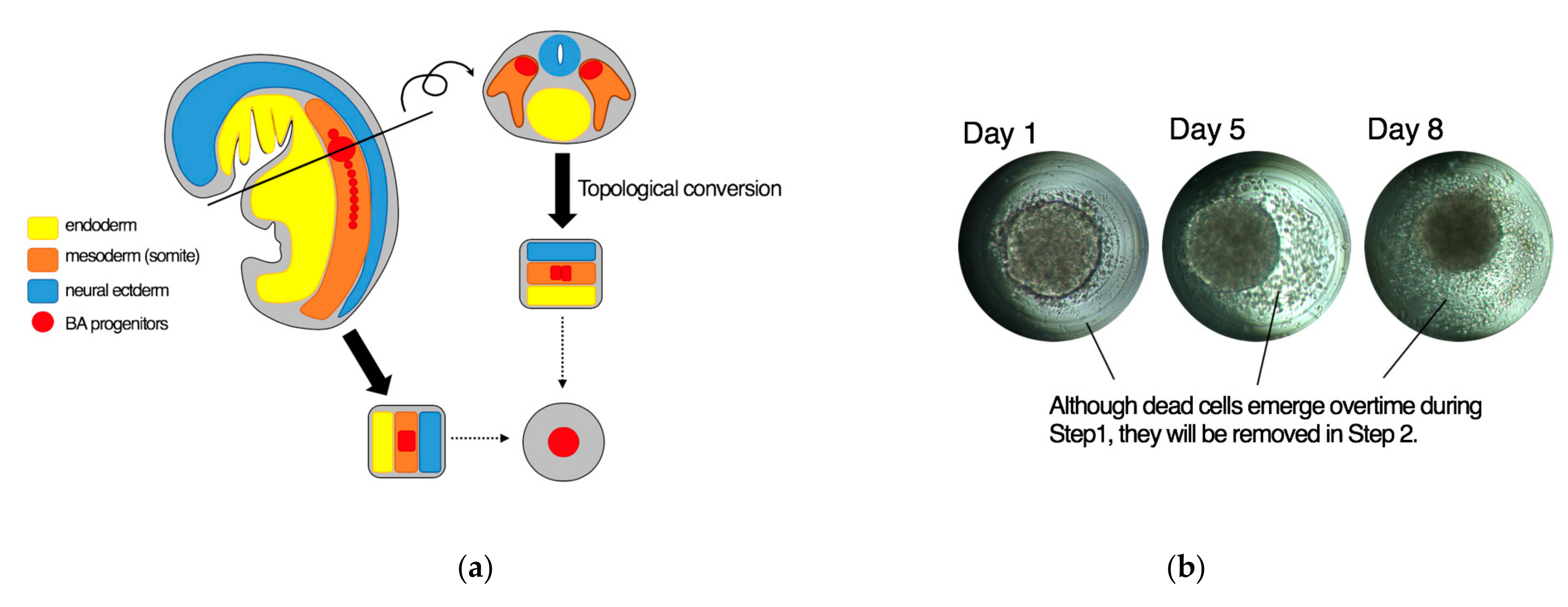
3.1. Adaptation of Human Pluripotent Stem Cells to a Feeder-Free Culture and an Induction of Differentiation
3.2. Culture of Size-Controlled Spheroids in Microwells Eliminates the Need for Exogenous Cytokines
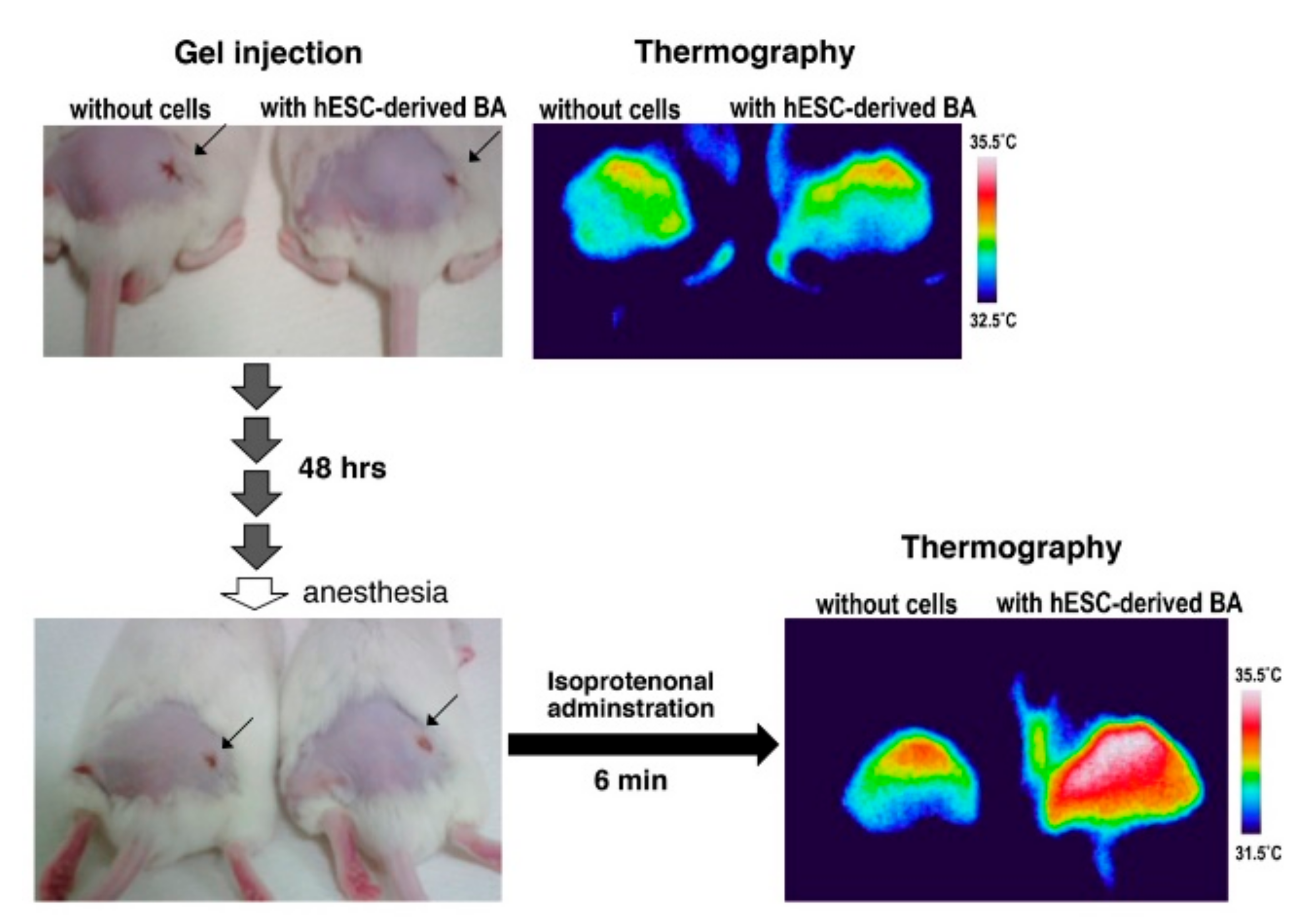
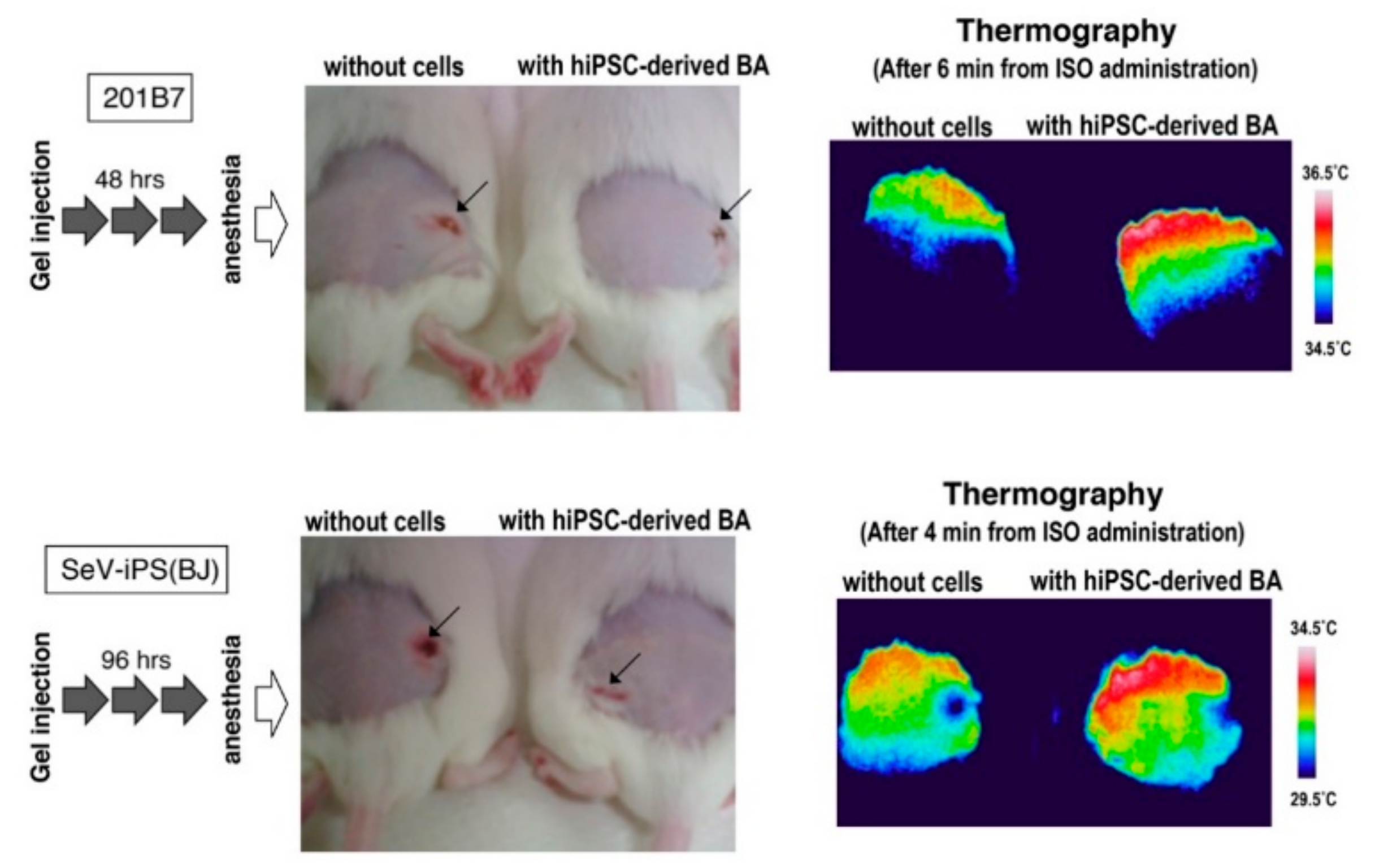
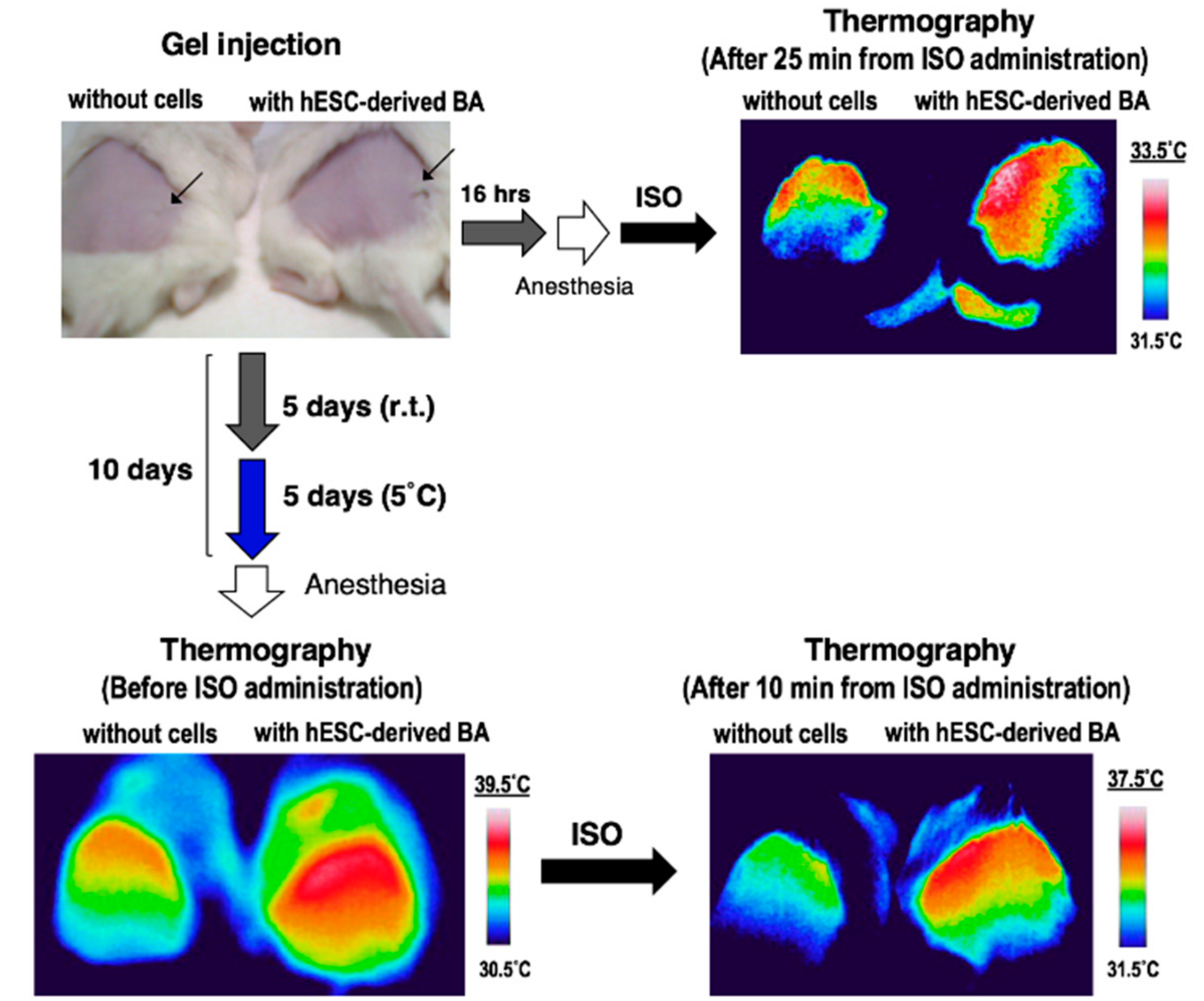
3.3. BA Produced Under an Exogenous Cytokine-Free Condition Exerts Thermogenic Activity In Vivo
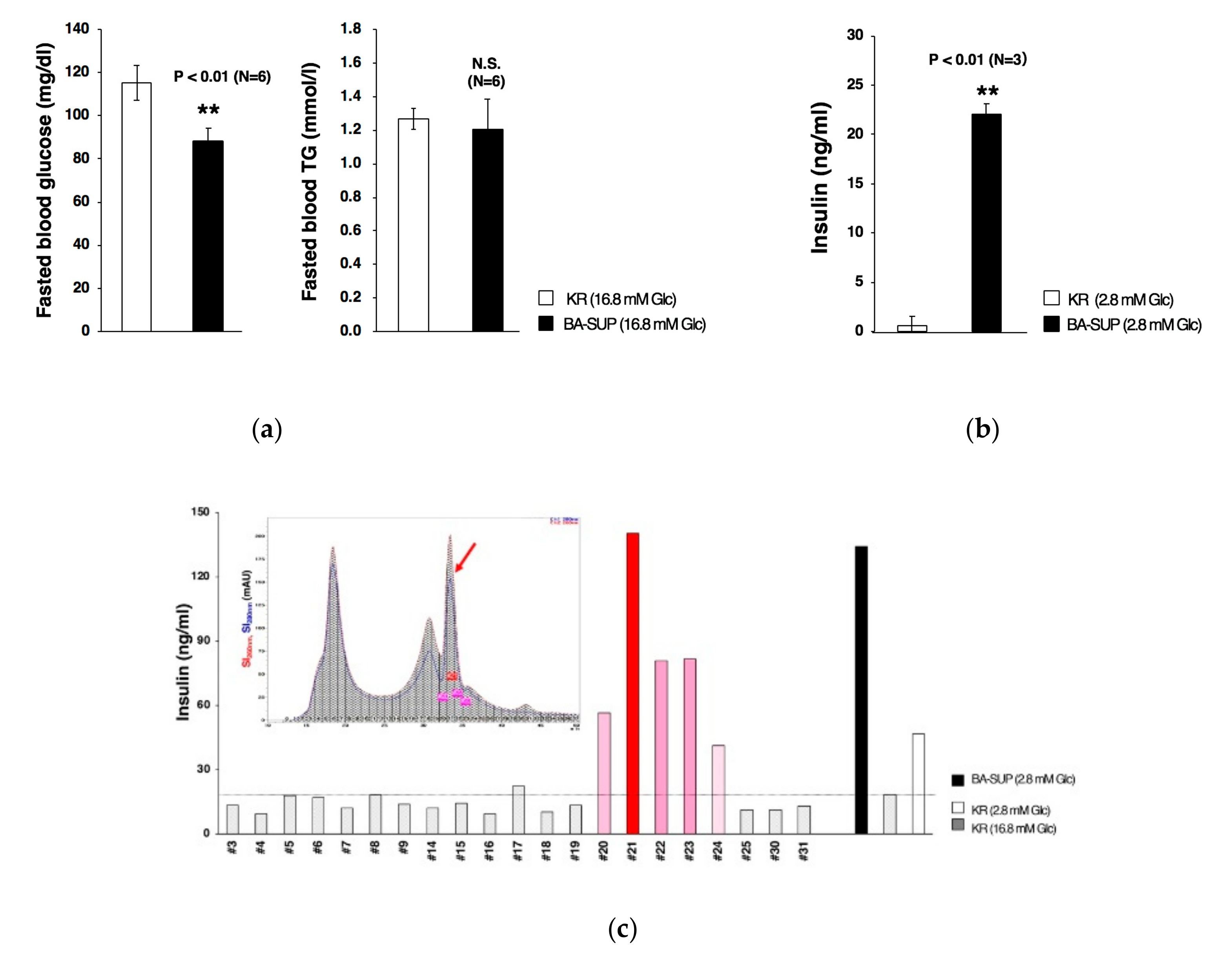
3.4. Partial Purification of Insulin Secretion-Enhancing Molecule from Supernatant of BA Produced Under an Exogenous Cytokine-Free Conditions
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guhr, A.; Kobold, S.; Seltmann, S.; Seiler Wulczyn, A.E.M.; Kurtz, A.; Löser, P. Recent Trends in Research with Human Pluripotent Stem Cells: Impact of Research and Use of Cell Lines in Experimental Research and Clinical Trials. Stem Cell Rep. 2018, 11, 485–496. [Google Scholar] [CrossRef]
- Galat, Y.; Dambaeva, S.; Elcheva, I.; Khanolkar, A.; Beaman, K.; Iannaccone, P.M.; Galat, V. Cytokine-free directed differentiation of human pluripotent stem cells efficiently produces hemogenic endothelium with lymphoid potential. Stem Cell Res. Ther. 2017, 8. [Google Scholar] [CrossRef]
- Stanford, K.I.; Middelbeek, R.J.; Townsend, K.L.; An, D.; Nygaard, E.B.; Hitchcox, K.M.; Markan, K.R.; Nakano, K.; Hirshman, M.F.; Tseng, Y.H.; et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J. Clin. Investig. 2013, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Yoneshiro, T.; Nakahara, M.; Suzuki, S.; Saeki, K.; Hasegawa, M.; Kawai, Y.; Akutsu, H.; Umezawa, A.; Yasuda, K.; et al. Production of functional classical brown adipocytes from human pluripotent stem cells using specific hemopoietin cocktail without gene introduction. Cell Metab. 2012, 16, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Saeki, K. Differentiation of human pluripotent stem cells into highly functional classical brown adipocytes. Methods Enzymol. 2014, 537, 177–197. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimeè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Nakahara, M.; Yuo, A.; Saeki, K. Human pluripotent stem cells: Towards therapeutic development for the treatment of lifestyle diseases. World J. Stem Cells 2016, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Sacks, H.; Symonds, M.E. Anatomical locations of human brown adipose tissue: Functional relevance and implications in obesity and type 2 diabetes. Diabetes 2013, 62, 1783–1790. [Google Scholar] [CrossRef]
- Clerte, M.; Baron, D.M.; Brouckaert, P.; Ernande, L.; Raher, M.J.; Flynn, A.W.; Picard, M.H.; Bloch, K.D.; Buys, E.S.; Scherrer-Crosbie, M. Brown adipose tissue blood flow and mass in obesity: A contrast ultrasound study in mice. Am. J. Soc. Echocardiogr. 2013, 26, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Waldeén, T.B.; Hansen, I.R.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E19–E31. [Google Scholar] [CrossRef]
- De Jong, J.M.; Larsson, O.; Cannon, B.; Nedergaard, J. A stringent validation of mouse adipose tissue identity markers. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1085–E1105. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Zhang, F.; Hao, G.; Shao, M.; Nham, K.; An, Y.; Wang, Q.; Zhu, Y.; Kusminski, C.M.; Hassan, G.; Gupta, R.K.; et al. An Adipose Tissue Atlas: An Image-Guided Identification of Human-like BAT and Beige Depots in Rodents. Cell Metab. 2018, 27, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerbäck, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Nakahara, M.; Oka, M.; Saeki, K. Could “Brown Adipose Tissue Failure” be a Cause of Metabolic Syndrome? Med. Res. Arch. 2016, 4. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, M.; Saeki, K.; Nakamura, N.; Matsuyama, S.; Yogiashi, Y.K.Y. A Human embryonic stem cells with maintenance under a feeder-free and recombinant cytokine-free condition. Cloning Stem Cells 2009, 11, 5–18. [Google Scholar] [CrossRef]
- Suemori, H.; Yasuchika, K.; Hasegawa, K.; Fujioka, T.; Tsuneyoshi, N.N. Efficient establishment of human embryonic stem cell lines and long-term maintenance with stable karyotype by enzymatic bulk passage. Biochem. Biophys. Res. Commun. 2006, 345, 926–932. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yamato, E.; Tashiro, F.; Miyazaki, J. Microarray analysis of novel candidate genes responsible for glucose-stimulated insulin secretion in mouse pancreatic β cell line MIN6. PLoS ONE 2013, 8, e61211. [Google Scholar] [CrossRef]
- Miyazaki, J.; Araki, K.; Yamato, E.; Ikegami, H.; Asano, T.; Shibasaki, Y.; Oka, Y.; Yamamura, K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: Special reference to expression of glucose transporter isoforms. Endocrinology 1990, 127, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Cypess, A.M.; Chen, Y.C.; Sze, C.; Wang, K.; English, J.; Chan, O.; Holman, A.R.; Tal, I.; Palmer, M.R.; Kolodny, G.M.; et al. Cold but not sympathomimetics activates human brown adipose tissue in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, 10001–10005. [Google Scholar] [CrossRef]
- Broeders, E.P.; Nascimento, E.B.; Havekes, B.; Brans, B.; Roumans, K.H.; Tailleux, A.; Schaart, G.; Kouach, M.; Charton, J.; Deprez, B.; et al. The Bile Acid Chenodeoxycholic Acid Increases Human Brown Adipose Tissue Activity. Cell Metab. 2015, 22, 418–426. [Google Scholar] [CrossRef]
- Atit, R.; Sgaier, S.K.; Mohamed, O.A.; Taketo, M.M.; Dufort, D.; Joyner, A.L.; Niswander, L.; Conlon, R.A. β-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev. Biol. 2006, 296, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Ogawa, T.; Okamoto, N.; Matsushita, M.; Aita, S.; Kameya, T.; Kawai, Y.; Iwanaga, T.; Saito, M. Impact of UCP1 and β3AR gene polymorphisms on age-related changes in brown adipose tissue and adiposity in humans. Int. J. Obes. 2013, 37, 993–998. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Original Method | Up-Graded Method | |
|---|---|---|
| Culture plate | any low attachment plates will do | plates that guarantee size-controlled spheroid formation |
| Cytokine cocktail | required | dispensable |
| Routine medium-changing procedure | easy and simple | requires labor and skill |
| Number of cells that can be handled at a time | large | small |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oka, M.; Kobayashi, N.; Matsumura, K.; Nishio, M.; Saeki, K. Exogenous Cytokine-Free Differentiation of Human Pluripotent Stem Cells into Classical Brown Adipocytes. Cells 2019, 8, 373. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8040373
Oka M, Kobayashi N, Matsumura K, Nishio M, Saeki K. Exogenous Cytokine-Free Differentiation of Human Pluripotent Stem Cells into Classical Brown Adipocytes. Cells. 2019; 8(4):373. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8040373
Chicago/Turabian StyleOka, Masako, Norihiko Kobayashi, Kazunori Matsumura, Miwako Nishio, and Kumiko Saeki. 2019. "Exogenous Cytokine-Free Differentiation of Human Pluripotent Stem Cells into Classical Brown Adipocytes" Cells 8, no. 4: 373. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8040373