The Aberrant Expression of the Mesenchymal Variant of FGFR2 in the Epithelial Context Inhibits Autophagy

 ,
,
Abstract
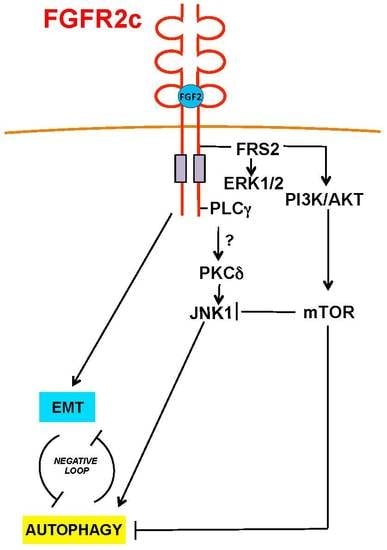
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. FGFR2c Expression and Signaling Inhibit Autophagy in Human Keratinocytes
2.2. The Autophagosome Formation is the Autophagic Step Impaired by FGFR2c Expression and Signaling
2.3. AKT/MTOR is the Downstream Pathway Responsible for FGFR2c-Mediated Inhibition of Autophagy
2.4. The Reversion of the Effects of FGFR2c on Autophagy Upon the Selective Block of AKT/MTOR Pathway is Accompanied by JNK1 Activation
2.5. Switching From the Epithelial FGFR2b to the Mesenchymal FGFR2c Isoform Underlies the FGF2-Mediated Inhibition of Autophagy in Epithelial Context
3. Discussion
4. Materials and Methods
4.1. Cells and Treatments
4.2. Immunoflurescence
4.3. Western Blot Analysis
4.4. Primers
4.5. RNA Extraction and cDNA Synthesis
4.6. PCR Amplification and Real-Time Quantitation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATG5 | autophagy-related 5 |
| BCL-2 | BCL2 apoptosis regulator |
| BECN1 | Beclin 1, autophagy-related |
| DAPI | 4′,6-diamidino-2-phenylindole |
| EGFP | enhanced green fluorescent protein |
| EMT | epithelial-mesenchymal transition |
| ESRP | Epithelial Splicing Regulatory Protein |
| FGF2 | fibroblast growth factor 2 (basic) |
| FGF7/KGF | fibroblast growth factor 7 |
| FGFR | fibroblast growth factor receptor |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HaCaT | human adult skin keratinocytes propagated under low calcium |
| HFs | Human fibroblasts |
| HKs | human keratinocytes |
| HPV16 | human papillomavirus type 16 |
| MAP1LC3/LC3 | microtubule-associated protein 1 light chain 3 |
| MAP2K/MEK | mitogen-activated protein kinase kinase |
| MAP2K4/MKK4 | mitogen-activated protein kinase kinase 4 |
| MAP2K7/MKK7 | mitogen-activated protein kinase kinase 7 |
| MAPK/ERK | mitogen-activated protein kinase |
| MAPK8/JNK | mitogen-activated protein kinase 8 |
| ACTB | actin beta |
| MAP3K5/ASK1 | mitogen-activated protein kinase kinase kinase 5 |
| MAPK8IP1/JIP1 | mitogen-activated protein kinase 8 interacting protein 1 |
| MLK | mixed-lineage kinase |
| MTOR | mechanistic target of rapamycin |
| NS | not significant |
| PI3K | class I phosphoinositide 3 kinase |
| PKC | protein kinase C |
| SD | standard deviation |
| SE | standard error |
| SDS-PAGE | sodium dodecyl sulphate-polyacrylamide gel electrophoresis |
| RT-PCR | Reverse Transcriptase-Polymerase Chain Reaction |
| siRNA | small interfering RNA |
| SQSTM1/P62 | sequestosome 1 |
| TUBA | α-tubulin |
References
- Brewer, J.R.; Mazot, P.; Soriano, P. Genetic insights into the mechanisms of FGF signaling. Genes Dev. 2016, 30, 751–771. [Google Scholar] [CrossRef] [PubMed]
- Tanner, Y.; Grose, R.P. Dysregulated FGF signalling in neoplastic disorders. Semin. Cell Dev. Biol. 2016, 53, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Brooks, A.N.; Kilgour, E.; Smith, P.D. Molecular pathways: Fibroblast growth factor signaling: A new therapeutic opportunity in cancer. Clin. Cancer Res. 2012, 18, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Conti, F.J.; Grose, R.; Revest, J.M.; Hodivala-Dilke, K.M.; Dickson, C. A crucial role for FGFR-IIIb signalling in epidermal development and hair follicle patterning. Development 2003, 130, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Grose, R.; Fantl, V.; Werner, S.; Chioni, A.M.; Jarosz, M.; Rudling, R.; Cross, B.; Hart, I.R.; Dickson, C. The role of fibroblast growth factor receptor 2b in skin homeostasis and cancer development. EMBO J. 2007, 26, 1268–1278. [Google Scholar] [CrossRef]
- Yang, J.; Meyer, M.; Müller, A.K.; Böhm, F.; Grose, R.; Dauwalder, T.; Verrey, F.; Kopf, M.; Partanen, J.; Bloch, W. Fibroblast growth factor receptors 1 and 2 in keratinocytes control the epidermal barrier and cutaneous homeostasis. J. Cell Biol. 2010, 188, 935–952. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Wang, F.; Matsubara, A.; Kan, M.; McKeehan, W.L. Fibroblast growth factor receptor 2 limits and receptor 1 accelerates tumorigenicity of prostate epithelial cells. Cancer Res. 1997, 58, 1509–1514. [Google Scholar]
- Zhang, Y.; Wang, H.; Toratani, S.; Sato, J.D.; Kan, M.; McKeehan, W.L.; Okamoto, T. Growth inhibition by keratinocyte growth factor receptor of human salivary adenocarcinoma cells through induction of differentiation and apoptosis. Proc. Natl. Acad. Sci. USA. 2001, 98, 11336–11340. [Google Scholar] [CrossRef] [Green Version]
- Belleudi, F.; Purpura, V.; Torrisi, M.R. The receptor tyrosine kinase FGFR2b/KGFR controls early differentiation of human keratinocytes. PLoS ONE 2011, 6, e24194. [Google Scholar] [CrossRef]
- Purpura, V.; Belleudi, F.; Caputo, S.; Torrisi, M.R. HPV16 E5 and KGFR/ FGFR2b interplay in differentiating epithelial cells. Oncotarget 2013, 4, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Rosato, B.; Ranieri, D.; Nanni, M.; Torrisi, M.R.; Belleudi, F. Role of FGFR2b expression and signaling in keratinocyte differentiation: Sequential involvement of PKCδ and PKCα. Cell Death Dis. 2018, 9, 565. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, D.; Rosato, B.; Nanni, M.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2c mesenchymal splicing variant in human keratinocytes inhibits differentiation and promotes invasion. Mol. Carcinog. 2018, 57, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, D.; Rosato, B.; Nanni, M.; Magenta, A.; Belleudi, F.; Torrisi, M.R. Expression of the FGFR2 mesenchymal splicing variant in epithelial cells drives epithelial-mesenchymal transition. Oncotarget 2016, 7, 5440–5460. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, D.; Belleudi, F.; Magenta, A.; Torrisi, M.R. HPV16 E5 expression induces switching from FGFR2b to FGFR2c and epithelial-mesenchymal transition. Int. J. Cancer 2015, 137, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Huang, Y.; Chang, J.Y.F.; Liu, L.; McKeehan, W.L.; Martin, J.F.; Wang, F. FRS2α-mediated FGF signals suppress premature differentiation of cardiac stem cells through regulating autophagy activity. Circ. Res. 2012, 110, e29–e39. [Google Scholar] [CrossRef] [PubMed]
- Cinque, L.; Forrester, A.; Bartolomeo, R.; Svelto, M.; Venditti, R.; Montefusco, S.; Polishchuk, E.; Nusco, E.; Rossi, A.; Medina, D. FGF signalling regulates bone growth through autophagy. Nature 2015, 528, 272–275. [Google Scholar] [CrossRef]
- Wang, X.; Qi, H.; Wang, Q.; Zhu, Y.; Wang, X.; Jin, M.; Tan, Q.; Huang, Q.; Xu, W.; Li, X. FGFR3/fibroblast growth factor receptor 3 inhibits autophagy through decreasing the ATG12-ATG5 conjugate, leading to the delay of cartilage development in achondroplasia. Autophagy 2015, 11, 1998–2013. [Google Scholar] [CrossRef]
- Belleudi, F.; Purpura, V.; Caputo, S.; Torrisi, M.R. FGF7/KGF regulates autophagy in keratinocytes: A novel dual role in the induction of both assembly and turnover of autophagosomes. Autophagy 2014, 10, 803–821. [Google Scholar] [CrossRef]
- Belleudi, F.; Nanni, M.; Raffa, S.; Torrisi, M.R. HPV16 E5 deregulates the autophagic process in human keratinocytes. Oncotarget 2015, 6, 9370–9386. [Google Scholar] [CrossRef] [Green Version]
- Nanni, M.; Ranieri, D.; Rosato, B.; Torrisi, M.R.; Belleudi, F. Role of fibroblast growth factor receptor 2b in the cross talk between autophagy and differentiation: Involvement of Jun N-terminal protein kinase signaling. Mol. Cell. Biol. 2018, 38, e00119-18. [Google Scholar]
- Nanni, M.; Ranieri, D.; Raffa, S.; Torrisi, M.R.; Belleudi, F. Interplay between FGFR2b-induced autophagy and phagocytosis: Role of PLCγ-mediated signaling. J. Cell Mol. Med. 2018, 22, 668–683. [Google Scholar] [CrossRef] [PubMed]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Ghezzi, P.; Rumio, C. The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol. Cancer 2017, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Rumio, C. How tumor cells choose between epithelial-mesenchymal transition and autophagy to resist stress-therapeutic implications. Front. Pharmacol. 2018, 9, 714. [Google Scholar] [CrossRef] [PubMed]
- Füllgrabe, J.; Ghislat, G.; Cho, D.H.; Rubinszteinet, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/ SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Zhang, Y.; Liu, L.; McKeehan, W.L.; Shen, Y.; Song, S.; Wang, F. FRS2α is essential for the fibroblast growth factor to regulate the MTOR pathway and autophagy in mouse embryonic fibroblasts. Int. J. Biol. Sci. 2011, 7, 1114–1121. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, X.; Li, X.; Wang, P.; Jing, Q.; Yue, J.; Liu, Y.; Cheng, Z.; Li, J.; Song, H. FGFR antagonist induces protective autophagy in FGFR1-amplified breast cancer cell. Biochem. Biophys. Res. Commun. 2016, 474, 1–7. [Google Scholar] [CrossRef]
- Yuan, H.; Li, Z.M.; Shao, J.; Ji, W.X.; Xia, W.; Lu, S. FGF2/FGFR1 regulates autophagy in FGFR1-amplified non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 72. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Mizuno, H.; Sase, H.; Fujii, T.; Sakata, K.; Akiyama, N.; Aoki, Y.; Aoki, M.; Ishii, N. ERK signal suppression and sensitivity to CH5183284/Debio 1347, a selective FGFR inhibitor. Mol. Cancer Ther. 2015, 14, 2831–2839. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Tony To, S.S. The phosphatidylinositol 3-kinase/Akt and c-Jun N-terminal kinase signaling in cancer: Alliance or contradiction? Int. J. Oncol. 2015, 47, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Blanco, J.; Martín, V.; García-Santos, G.; Herrera, F.; Casado-Zapico, S.; Antolín, I.; Rodriguez, C. Cooperative action of JNK and AKT/MTOR in 1-methyl-4-phenylpyridinium-induced autophagy of neuronal PC12 cells. J. Neurosci. Res. 2012, 90, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell. 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Sato, T.K.; Nabet, B.; Hogenesch, J.B.; Carstens, R.P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell. 2009, 33, 591–601. [Google Scholar] [CrossRef]
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458. [Google Scholar] [CrossRef]
- Kawase, R.; Ishiwata, T.; Matsuda, Y.; Onda, M.; Kudo, M.; Takeshita, T.; Naito, Z. Expression of fibroblast growth factor receptor 2 IIIc in human uterine cervical intraepithelial neoplasia and cervical cancer. Int. J. Oncol. 2010, 36, 331–340. [Google Scholar]
- Matsuda, Y.; Hagio, M.; Seya, T.; Ishiwata, T. Fibroblast growth factor receptor 2 IIIc as a therapeutic target for colorectal cancer cells. Mol. Cancer Ther. 2012, 11, 2010–2020. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, T.; Matsuda, Y.; Yamamoto, T.; Uchida, E.; Korc, M.; Naito, Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. Am. J. Pathol. 2012, 180, 1928–1941. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Caballero, O.L.; Davis, I.D.; Jonasch, E.; Tamboli, P.; Yung, W.K.; Weinstein, J.N.; Kenna Shaw for TCGA research network; Strausberg, R.L.; Yao, J. Tumor-specific isoform switch of the fibroblast growth factor receptor 2 underlies the mesenchymal and malignant phenotypes of clear cell renal cell carcinomas. Clin. Cancer Res. 2013, 19, 2460–2472. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Kudo, M.; Fujii, T.; Teduka, K.; Naito, Z. Altered expression of fibroblast growth factor receptor 2 isoform IIIc: Relevance to endometrioid adenocarcinoma carcinogenesis and histological differentiation. Int. J. Clin. Exp. Pathol. 2014, 7, 1069–1076. [Google Scholar] [PubMed]
- Urbanski, L.M.; Leclair, N.; Anczuków, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, L.; Zhong, J.; Wu, N.; Liu, G.; Lin, X. Oleanolic acid induces protective autophagy in cancer cells through the JNK and MTOR pathways. Oncol. Rep. 2014, 32, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Yano, H.; Cho, H.; Meyer, D.; Monks, B.; Margolis, B.; Birnbaum, M.J.; Chao, M.V. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 2002, 35, 697–709. [Google Scholar] [CrossRef]
- Pan, J.; Pei, D.S.; Yin, X.H.; Hui, L.; Zhang, G.Y. Involvement of oxidative stress in the rapid Akt1 regulating a JNK scaffold during ischemia in rat hippocampus. Neurosci. Lett. 2006, 392, 47–51. [Google Scholar] [CrossRef]
- Cardinali, G.; Ceccarelli, S.; Kovacs, D.; Aspite, N.; Lotti, L.V.; Torrisi, M.R.; Picardo, M. Keratinocyte growth factor promotes melanosome transfer to keratinocytes. J. Investig. Dermatol. 2005, 125, 1190–1199. [Google Scholar] [CrossRef]
- Raffa, S.; Leone, L.; Scrofani, C.; Monini, S.; Torrisi, M.R.; Barbara, M. Cholesteatoma-associated fibroblasts modulate epithelial growth and differentiation through KGF/FGF7 secretion. Histochem. Cell Biol. 2012, 138, 251–269. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, D.; Genovese, L.; Ponti, D.; Ranieri, D.; Raffa, S.; Calogero, A.; Torrisi, M.R. Nucleolar localization and circadian regulation of Per2S, a novel splicing variant of the Period 2gene. Cell Mol. Life Sci. 2014, 71, 2547–2559. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanni, M.; Ranieri, D.; Persechino, F.; Torrisi, M.R.; Belleudi, F. The Aberrant Expression of the Mesenchymal Variant of FGFR2 in the Epithelial Context Inhibits Autophagy. Cells 2019, 8, 653. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070653
Nanni M, Ranieri D, Persechino F, Torrisi MR, Belleudi F. The Aberrant Expression of the Mesenchymal Variant of FGFR2 in the Epithelial Context Inhibits Autophagy. Cells. 2019; 8(7):653. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070653
Chicago/Turabian StyleNanni, Monica, Danilo Ranieri, Flavia Persechino, Maria Rosaria Torrisi, and Francesca Belleudi. 2019. "The Aberrant Expression of the Mesenchymal Variant of FGFR2 in the Epithelial Context Inhibits Autophagy" Cells 8, no. 7: 653. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8070653