Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neurodegeneration Induced In Vitro by α-Synuclein Fibrillary Aggregates

 , , and
, , and 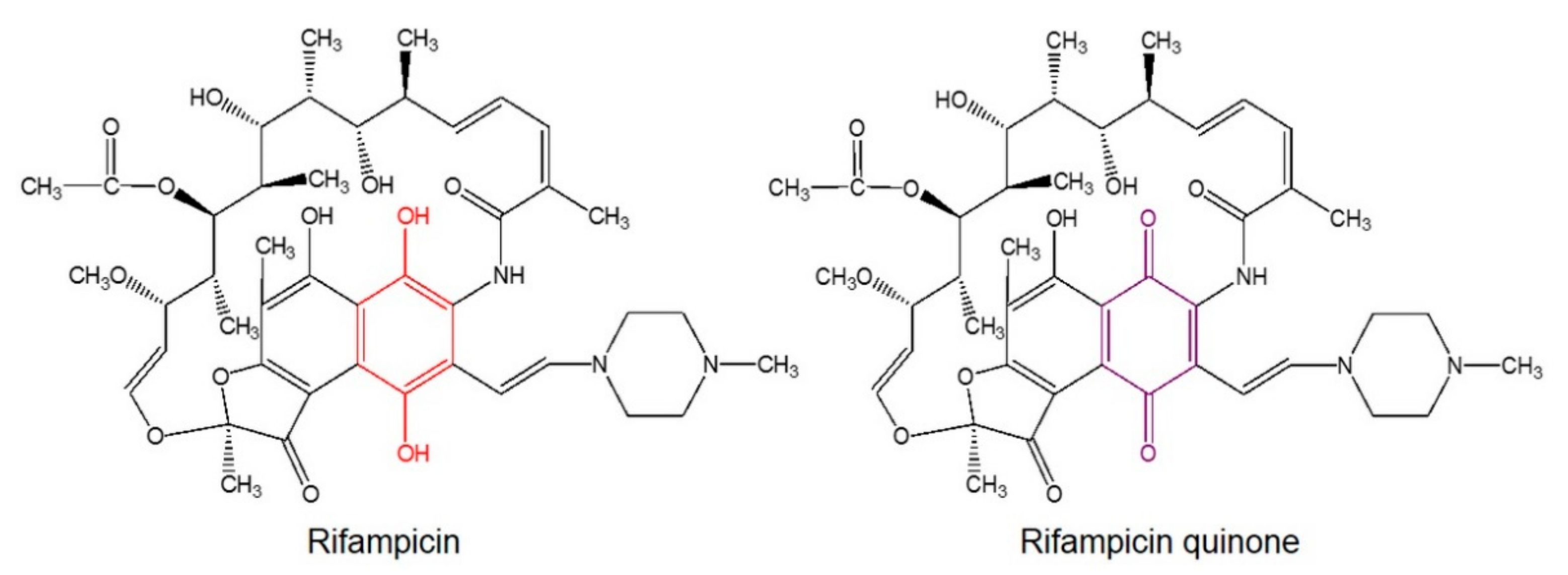{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of α-synuclein
2.2. Transmission Electron Microscopy
2.3. Cell Culture Protocols
2.3.1. Microglial Cell Isolation
2.3.2. Microglial Cell Stimulation and Treatments
2.3.3. Primary Cortical Neuron Cultures
2.3.4. Neurotoxicity Assays
2.3.5. Microglial-Conditioned Media Preparation
2.4. Protein Detection by Immunofluorescence
2.5. Western Blot Analysis
2.6. NADPH Oxidase Activity
2.7. Statistical Analysis
3. Results
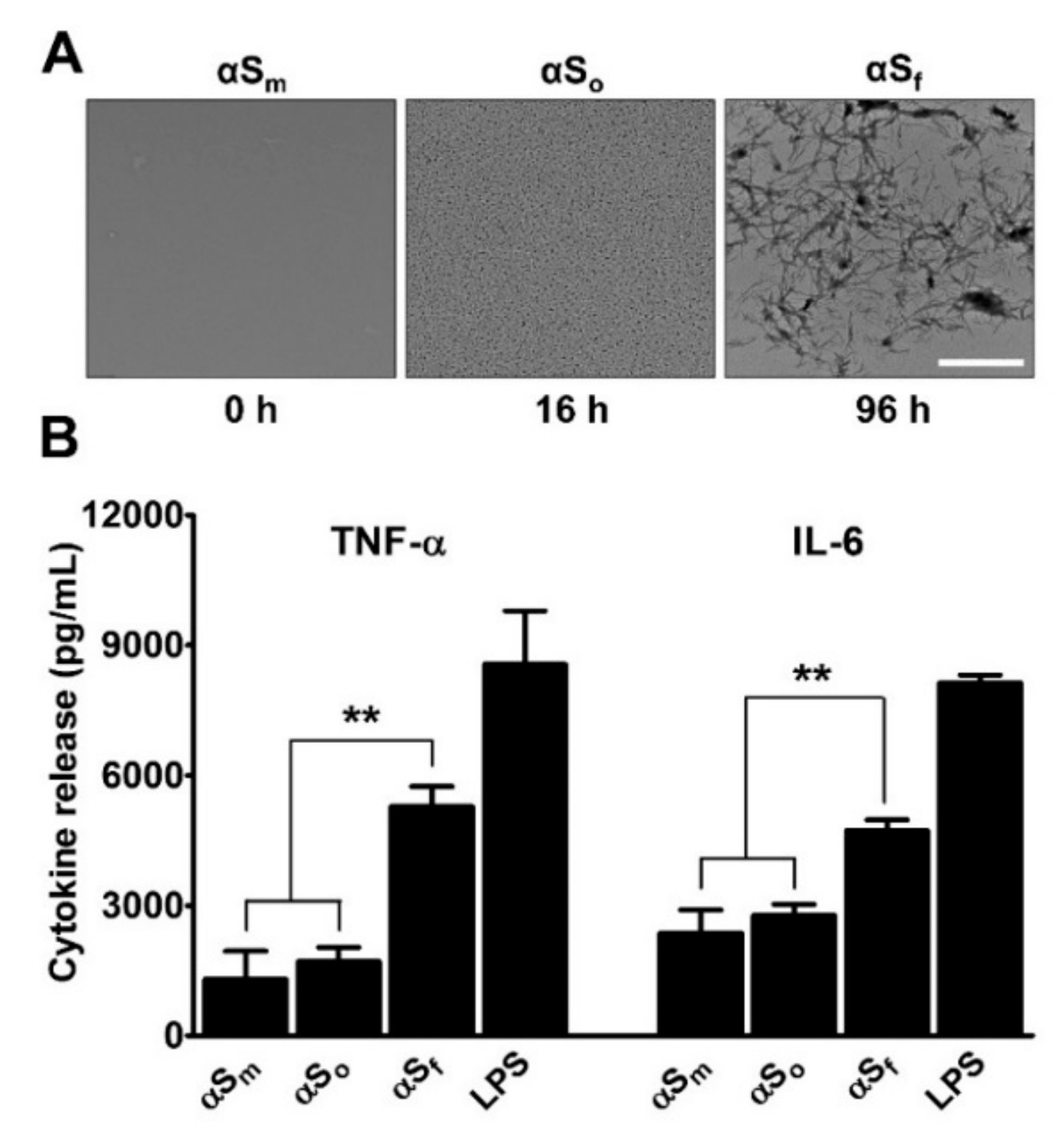
3.1. α-Synuclein Fibrils Induce Pro-Inflammatory Cytokine Release in Microglial Cell Cultures
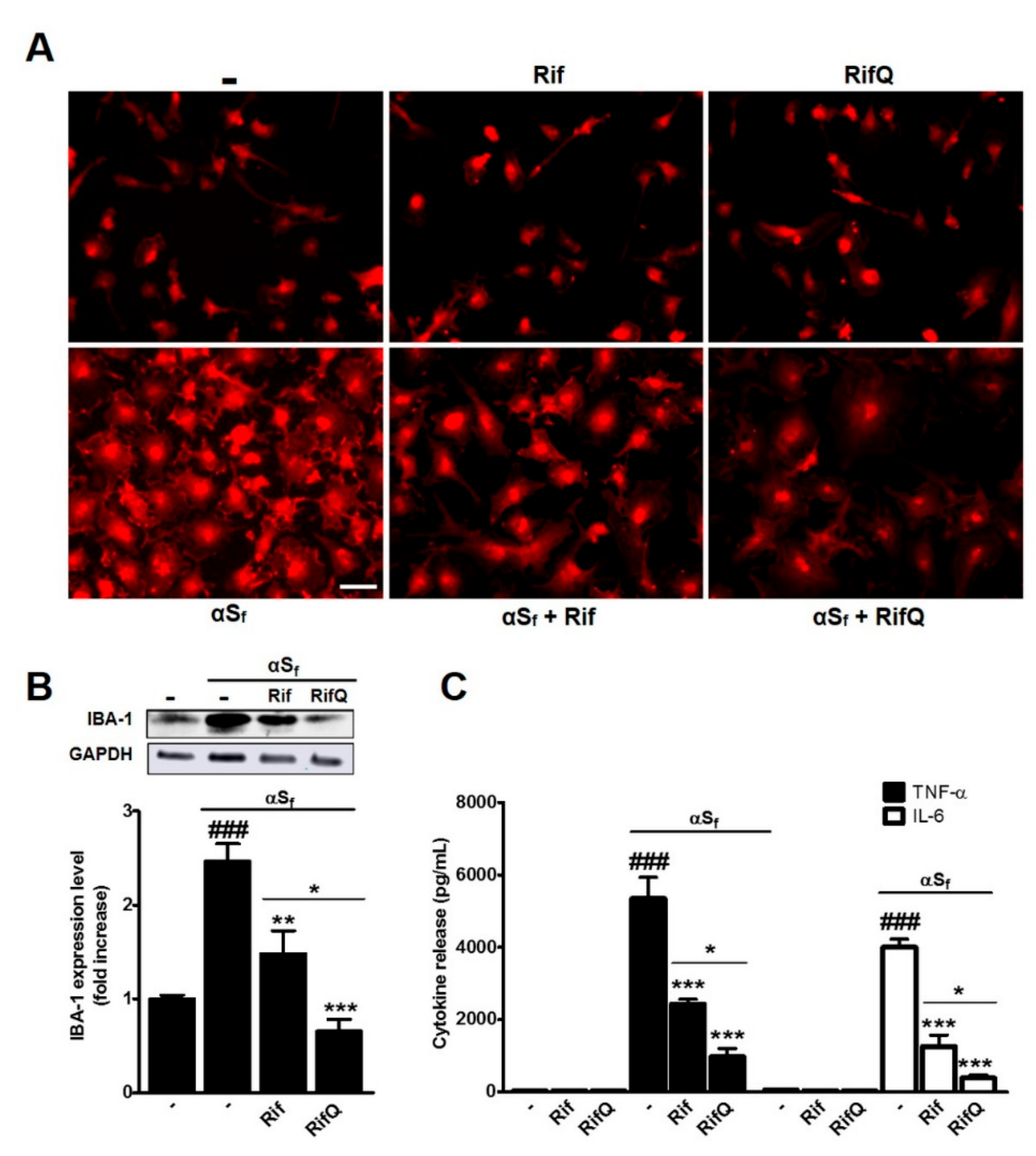
3.2. Rifampicin and Rifampicin Quinone Prevent Microglial Activation Induced by α-Synuclein Fibrils
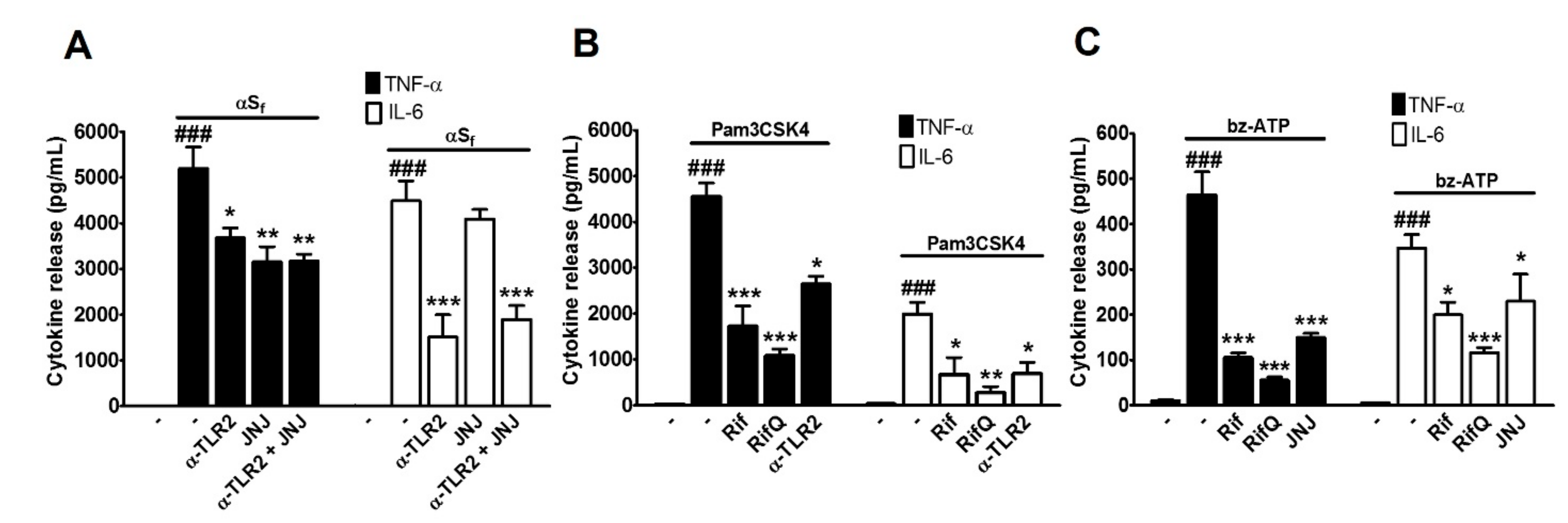
3.3. Rifampicin and Rifampicin Quinone Prevent TLR2- and P2X7-Dependent Microglial Activation
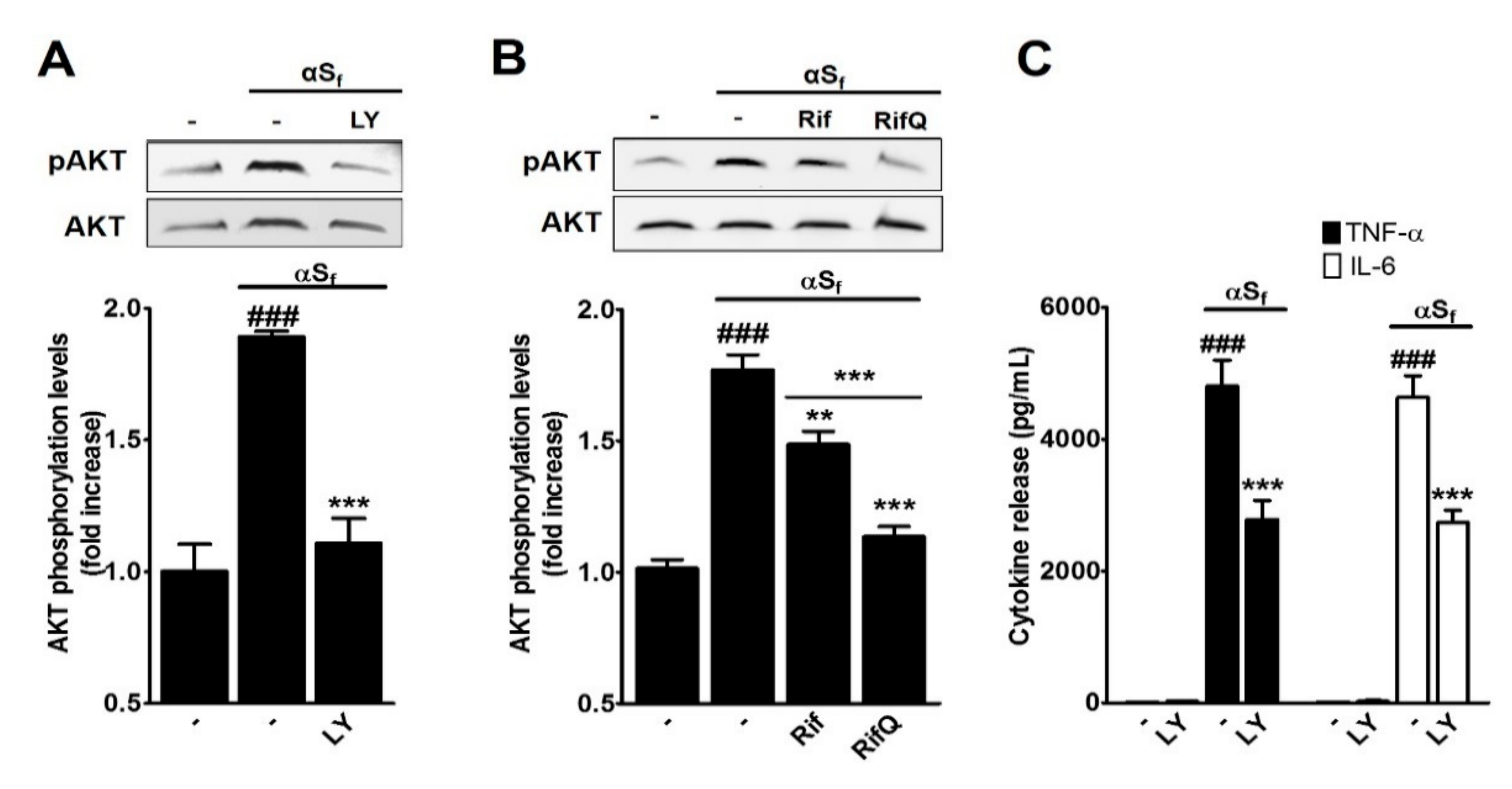
3.4. Inhibitory Effect of Rif and RifQ on αSf-Induced PI3K/AKT Activity
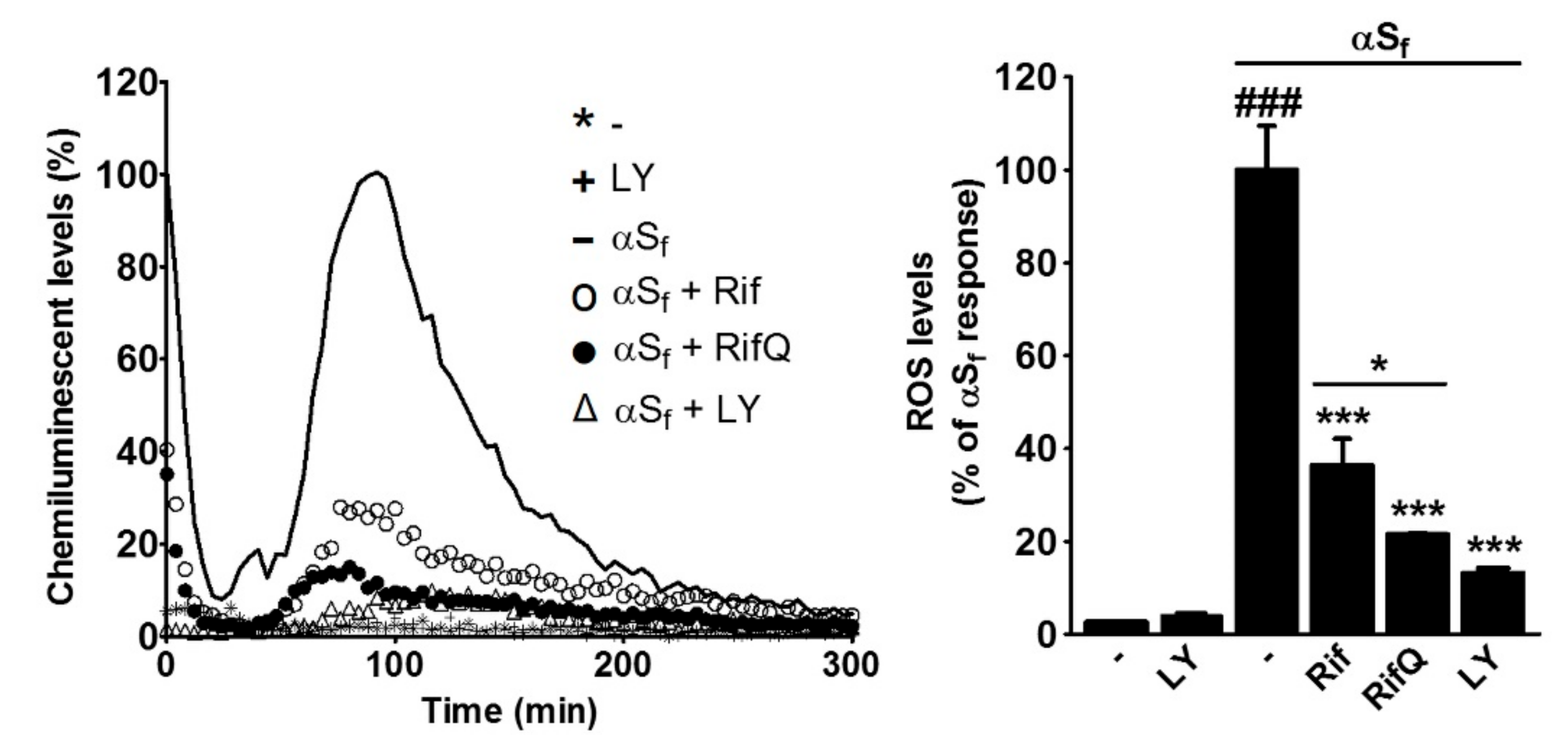
3.5. Rifampicin and Rifampicin Quinone Prevent Reactive Oxygen Species Production in Microglial Cells Activated by αSf
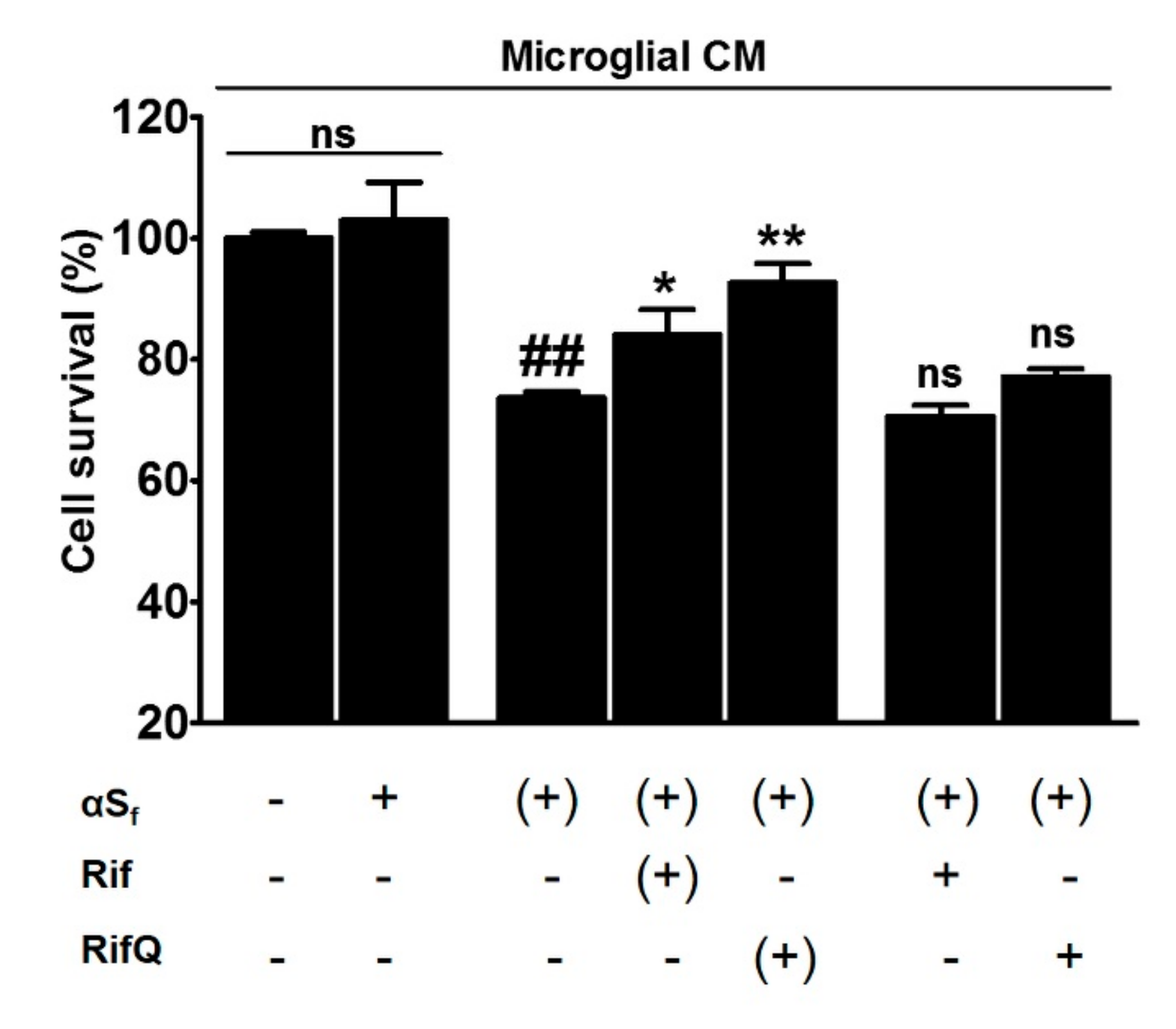
3.6. Rifampicin Quinone Protects Cortical Neurons Against Death Caused by αSf-Induced Microglial Activation
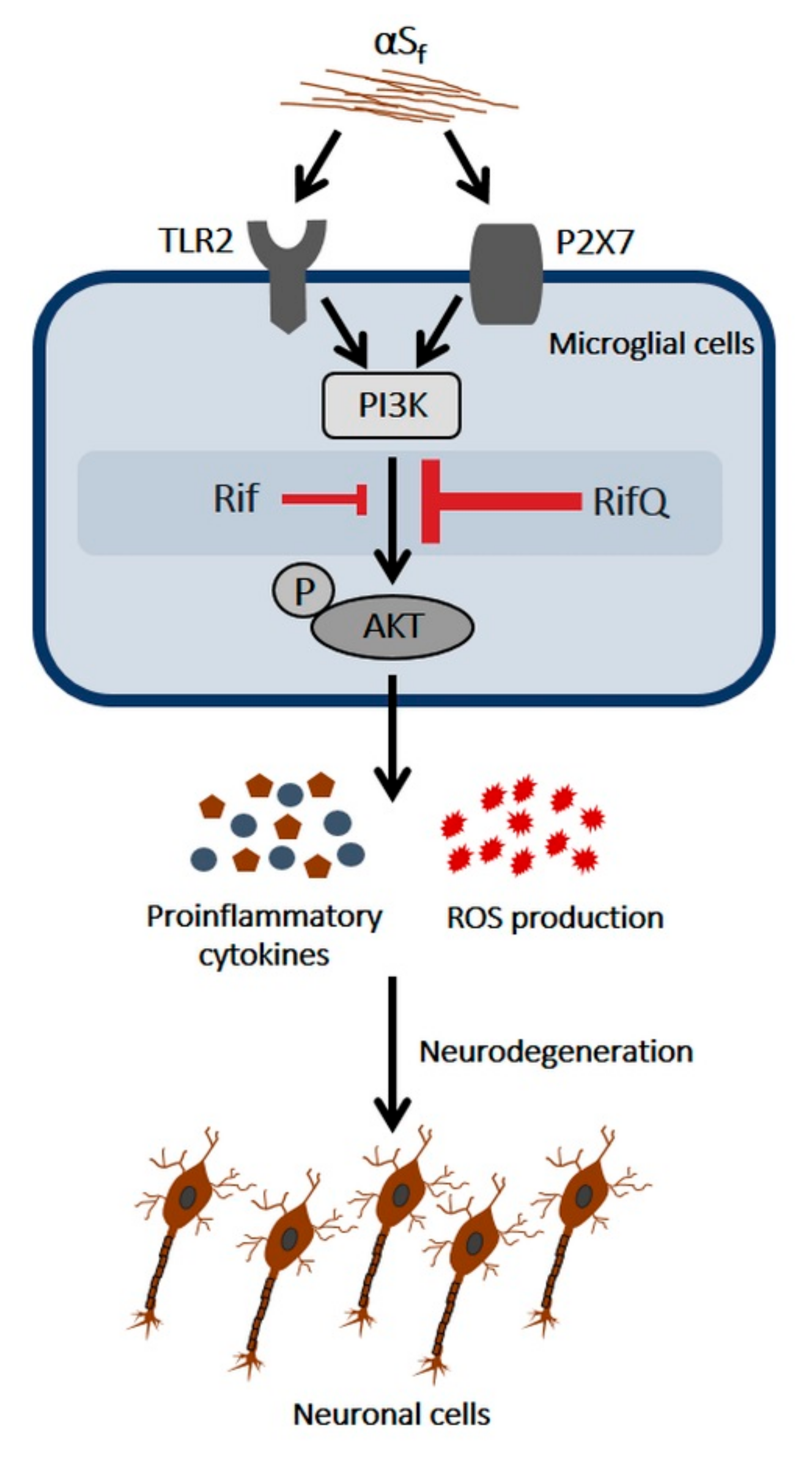
4. Discussion
4.1. Fibrillary Aggregates of αS are the Most Inflammogenic Forms of αS for Microglial Cells
4.2. RifQ is More Efficient than Rif in Reducing αSf-Induced Microglial Cell Activation
4.3. Rif and RifQ Inhibit an Activation Process Mediated by TLR2 and P2X7 Receptors
4.4. Rifampicin and Rifampicin Quinone act as Inhibitors of PI3K/AKT-Dependent Signaling
4.5. Rifampicin Quinone Provides Neuroprotection through its Anti-Inflammatory Activity
Author Contributions
Funding
Conflicts of Interest
References
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The role of alpha-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Bae, E.-J.; Lee, S.-J. Extracellular α-Snuclein-a novel and crucial factor in Lewy body diseases. Nat. Rev. Neurol. 2014, 10, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Akiyama, H.; McGeer, E.G. Rate of cell death in parkinsonism indicates active neuropathological process. Ann. Neurol. 1988, 24, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Iseki, E.; Marui, W.; Akiyama, H.; Uéda, K.; Kosaka, K. Degeneration process of Lewy bodies in the brains of patients with dementia with Lewy bodies using alpha-synuclein-immunohistochemistry. Neurosci. Lett. 2000, 286, 69–73. [Google Scholar] [CrossRef]
- Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience 2015, 302, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Yulug, B.; Hanoglu, L.; Kilic, E.; Schabitz, W.R. RIFAMPICIN: An antibiotic with brain protective function. Brain Res. Bull. 2014, 107, 37–42. [Google Scholar] [CrossRef]
- González-Lizárraga, F.; Socías, S.B.; Ávila, C.L.; Torres-Bugeau, C.M.; Barbosa, L.R.S.; Binolfi, A.; Sepúlveda-Díaz, J.E.; del-Bel, E.; Fernandez, C.O.; Papy-Garcia, D.; et al. Repurposing doxycycline for synucleinopathies: Remodelling of α-synuclein oligomers towards non-toxic parallel beta-sheet structured species. Sci. Rep. 2017, 7, 41755. [Google Scholar] [CrossRef]
- Forloni, G.; Artuso, V.; Roiter, I.; Morbin, M.; Tagliavini, F. Therapy in prion diseases. Curr. Top. Med. Chem. 2013, 13, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Stoilova, T.; Colombo, L.; Forloni, G.; Tagliavini, F.; Salmona, M. A new face for old antibiotics: Tetracyclines in treatment of amyloidoses. J. Med. Chem. 2013, 56, 5987–6006. [Google Scholar] [CrossRef] [PubMed]
- Santa-Cecília, F.V.; Socias, B.; Ouidja, M.O.; Sepulveda-Diaz, J.E.; Acuña, L.; Silva, R.L.; Michel, P.P.; Del-Bel, E.; Cunha, T.M.; Raisman-Vozari, R. Doxycycline Suppresses Microglial Activation by Inhibiting the p38 MAPK and NF-kB Signaling Pathways. Neurotox. Res. 2016, 29, 447–459. [Google Scholar] [CrossRef]
- Uppal, L.; Singhi, S.; Singhi, P.; Aggarwal, R. Role of Rifampin in Reducing Inflammation and Neuronal Damage in Childhood Bacterial Meningitis: A Pilot Randomized Controlled Trial. Pediatr. Infect. Dis. J. 2017, 36, 556–559. [Google Scholar] [CrossRef]
- McGeer, P.L.; Harada, N.; Kimura, H.; McGeer, E.G.; Schulzer, M. Prevalence of Dementia amongst Elderly Japanese with Leprosy: Apparent Effect of Chronic Drug Therapy. Dement. Geriatr. Cognit. Disord. 1992, 3, 146–149. [Google Scholar] [CrossRef]
- Chui, D.H.; Tabira, T.; Izumi, S.; Koya, G.; Ogata, J. Decreased beta-amyloid and increased abnormal Tau deposition in the brain of aged patients with leprosy. Am. J. Pathol. 1994, 145, 771–775. [Google Scholar] [PubMed]
- Bi, W.; Zhu, L.; Jing, X.; Zeng, Z.; Liang, Y.; Xu, A.; Liu, J.; Xiao, S.; Yang, L.; Shi, Q.; et al. Rifampicin improves neuronal apoptosis in LPS-stimulated co-cultured BV2 cells through inhibition of the TLR-4 pathway. Mol. Med. Rep. 2014, 10, 1793–1799. [Google Scholar] [CrossRef]
- Bi, W.; Zhu, L.; Wang, C.; Liang, Y.; Liu, J.; Shi, Q.; Tao, E. Rifampicin inhibits microglial inflammation and improves neuron survival against inflammation. Brain Res. 2011, 1395, 12–20. [Google Scholar] [CrossRef]
- Kilic, U.; Kilic, E.; Lingor, P.; Yulug, B.; Bähr, M. Rifampicin inhibits neurodegeneration in the optic nerve transection model in vivo and after 1-methyl-4-phenylpyridinium intoxication in vitro. Acta Neuropathol. 2004, 108, 65–68. [Google Scholar] [CrossRef]
- Liang, Y.; Jing, X.; Zeng, Z.; Bi, W.; Chen, Y.; Wu, X.; Yang, L.; Liu, J.; Xiao, S.; Liu, S.; et al. Rifampicin attenuates rotenone-induced inflammation via suppressing NLRP3 inflammasome activation in microglia. Brain Res. 2015, 1622, 43–50. [Google Scholar] [CrossRef]
- Li, J.; Zhu, M.; Rajamani, S.; Uversky, V.N.; Fink, A.L. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem. Biol. 2004, 11, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Konrad, P.; Stenberg, P. Rifampicin quinone is an immunosuppressant, but not rifampicin itself. Clin. Immunol. Immunopathol. 1988, 46, 162–166. [Google Scholar] [CrossRef]
- Sepulveda-Diaz, J.E.; Ouidja, M.O.; Socias, S.B.; Hamadat, S.; Guerreiro, S.; Raisman-Vozari, R.; Michel, P.P. A simplified approach for efficient isolation of functional microglial cells: Application for modeling neuroinflammatory responses in vitro. Glia 2016, 64, 1912–1924. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef]
- Dos-Santos-Pereira, M.; Acuña, L.; Hamadat, S.; Rocca, J.; González-Lizárraga, F.; Chehín, R.; Sepulveda-Diaz, J.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Microglial glutamate release evoked by α-synuclein aggregates is prevented by dopamine. Glia 2018, 66, 2353–2365. [Google Scholar] [CrossRef] [PubMed]
- Fifre, A.; Sponne, I.; Koziel, V.; Kriem, B.; Potin, F.T.Y.; Bihain, B.E.; Olivier, J.-L.; Oster, T.; Pillot, T. Microtubule-associated Protein MAP1A, MAP1B, and MAP2 Proteolysis during Soluble Amyloid β-Peptide-induced Neuronal Apoptosis Synergistic Involvement of Calpain and Caspase-3. J. Biol. Chem. 2006, 281, 229–240. [Google Scholar] [CrossRef] [PubMed]
- LeVine, H. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999, 309, 274–284. [Google Scholar] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Ito, D.; Tanaka, K.; Suzuki, S.; Dembo, T.; Fukuuchi, Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 2001, 32, 1208–1215. [Google Scholar] [CrossRef]
- Singh, V.; Mitra, S.; Sharma, A.K.; Gera, R.; Ghosh, D. Isolation and characterization of microglia from adult mouse brain: Selected applications for ex vivo evaluation of immunotoxicological alterations following in vivo xenobiotic exposure. Chem. Res. Toxicol. 2014, 27, 895–903. [Google Scholar] [CrossRef]
- St Paul, M.; Barjesteh, N.; Paolucci, S.; Pei, Y.; Sharif, S. Toll-like receptor ligands induce the expression of interferon-gamma and interleukin-17 in chicken CD4+ T cells. BMC Res. Notes 2012, 5, 616. [Google Scholar] [CrossRef] [PubMed]
- Young, M.T.; Pelegrin, P.; Surprenant, A. Amino acid residues in the P2X7 receptor that mediate differential sensitivity to ATP and BzATP. Mol. Pharmacol. 2007, 71, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Grabiec, A.; Rutz, M.; Metzger, J.; Luppa, P.B.; Wagner, H.; Bauer, S.; Kirschning, C.J. Murine TLR2 expression analysis and systemic antagonism by usage of specific monoclonal antibodies. Immunol. Lett. 2005, 98, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Letavic, M.A.; Lord, B.; Bischoff, F.; Hawryluk, N.A.; Pieters, S.; Rech, J.C.; Sales, Z.; Velter, A.I.; Ao, H.; Bonaventure, P.; et al. Synthesis and Pharmacological Characterization of Two Novel, Brain Penetrating P2X7 Antagonists. ACS Med. Chem. Lett. 2013, 4, 419–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Hoekstra, J.; Heng, X.; Kang, W.; Ding, J.; Liu, J.; Chen, S.; Zhang, J. P2X7 receptor is critical in α-synuclein—Mediated microglial NADPH oxidase activation. Neurobiol. Aging 2015, 36, 2304–2318. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Tang, C.-H.; Chen, Y.-H.; Wei, I.-H.; Chen, J.-H.; Lai, C.-H.; Lu, D.-Y. Peptidoglycan enhances proinflammatory cytokine expression through the TLR2 receptor, MyD88, phosphatidylinositol 3-kinase/AKT and NF-kappaB pathways in BV-2 microglia. Int. Immunopharmacol. 2010, 10, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, C.; Cianciulli, A.; Calvello, R.; Dragone, T.; Iacobazzi, F.; Panaro, M.A. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharmacol. Immunotoxicol. 2012, 34, 858–865. [Google Scholar] [CrossRef]
- Vergara, D.; Nigro, A.; Romano, A.; de Domenico, S.; Damato, M.; Franck, J.; Coricciati, C.; Wistorski, M.; Cardon, T.; Fournier, I.; et al. Distinct Protein Expression Networks are Activated in Microglia Cells after Stimulation with IFN-γ and IL-4. Cells 2019, 8, 580. [Google Scholar] [CrossRef]
- Bernis, M.E.; Babila, J.T.; Breid, S.; Wüsten, K.A.; Wüllner, U.; Tamgüney, G. Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol. Commun. 2015, 3, 75. [Google Scholar] [CrossRef]
- Dijkstra, A.A.; Voorn, P.; Berendse, H.W.; Groenewegen, H.J.; Netherlands Brain Bank; Rozemuller, A.J.M.; van de Berg, W.D.J. Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1244–1251. [Google Scholar] [CrossRef]
- Couch, Y.; Alvarez-Erviti, L.; Sibson, N.R.; Wood, M.J.A.; Anthony, D.C. The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J. Neuroinflamm. 2011, 8, 166. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.-C.; von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C.M. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Gustot, A.; Gallea, J.I.; Sarroukh, R.; Celej, M.S.; Ruysschaert, J.-M.; Raussens, V. Amyloid fibrils are the molecular trigger of inflammation in Parkinson’s disease. Biochem. J. 2015, 471, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Chavarría, C.; Rodríguez-Bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.; Zhu, L.; Jing, X.; Liang, Y.; Tao, E. Rifampicin and Parkinson’s disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2013, 34, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.D.; Choi, M.L.; Ryten, M.; Hopkins, L.; Drews, A.; Botía, J.A.; Iljina, M.; Rodrigues, M.; Gagliano, S.A.; Gandhi, S.; et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019, 137, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.N.; Aperia, A.; Melki, R.; Triller, A. Physico-Pathologic Mechanisms Involved in Neurodegeneration: Misfolded Protein-Plasma Membrane Interactions. Neuron 2017, 95, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.-F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Drouin-Ouellet, J.; St-Amour, I.; Saint-Pierre, M.; Lamontagne-Proulx, J.; Kriz, J.; Barker, R.A.; Cicchetti, F. Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int. J. Neuropsychopharmacol. 2014, 18. [Google Scholar] [CrossRef]
- Kim, C.; Lee, H.-J.; Masliah, E.; Lee, S.-J. Non-cell-autonomous Neurotoxicity of α-synuclein Through Microglial Toll-like Receptor 2. Exp. Neurobiol. 2016, 25, 113–119. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Cho, E.-D.; Kim, H.-K.; You, S.; Lee, H.-J.; Hwang, D.; Lee, S.-J. β1-integrin-dependent migration of microglia in response to neuron-released α-synuclein. Exp. Mol. Med. 2014, 46, e91. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.-H.; Yan, W.-F.; Zhang, Z.; Ma, K.-L.; Peng, S.-Y.; Cao, Y.-L.; Yuan, Y.-H.; Chen, N.-H. Nurr1: A vital participant in the TLR4-NF-κB signal pathway stimulated by α-synuclein in BV-2 cells. Neuropharmacology 2019, 144, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Ifuku, M.; Buonfiglioli, A.; Jordan, P.; Lehnardt, S.; Kettenmann, H. TLR2 controls random motility, while TLR7 regulates chemotaxis of microglial cells via distinct pathways. Brain. Behav. Immun. 2016, 58, 338–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, M.O.; Rosa, A.I.; Carvalho, A.N.; Nunes, M.J.; Dionísio, P.; Rodrigues, E.; Costa, D.; Duarte-Silva, S.; Maciel, P.; Rodrigues, C.M.P.; et al. Neurotoxic effects of MPTP on mouse cerebral cortex: Modulation of neuroinflammation as a neuroprotective strategy. Mol. Cell. Neurosci. 2019, 96, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Esparcia, P.; Koneti, A.; Rodríguez-Oroz, M.C.; Gago, B.; del Rio, J.A.; Ferrer, I. Mitochondrial activity in the frontal cortex area 8 and angular gyrus in Parkinson’s disease and Parkinson’s disease with dementia. Brain Pathol. 2018, 28, 43–57. [Google Scholar] [CrossRef]
- Foffani, G.; Obeso, J.A. A Cortical Pathogenic Theory of Parkinson’s Disease. Neuron 2018, 99, 1116–1128. [Google Scholar] [CrossRef]
- Falkenburger, B.H.; Saridaki, T.; Dinter, E. Cellular models for Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 121–130. [Google Scholar] [CrossRef]
- Bussi, C.; Ramos, J.M.P.; Arroyo, D.S.; Gaviglio, E.A.; Gallea, J.I.; Wang, J.M.; Celej, M.S.; Iribarren, P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci. Rep. 2017, 7, 43153. [Google Scholar] [CrossRef]
- Wang, S.; Chu, C.-H.; Guo, M.; Jiang, L.; Nie, H.; Zhang, W.; Wilson, B.; Yang, L.; Stewart, T.; Hong, J.-S.; et al. Identification of a specific α-synuclein peptide (α-Syn 29-40) capable of eliciting microglial superoxide production to damage dopaminergic neurons. J. Neuroinflamm. 2016, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wei, C.; Xu, C.; Bennett, M.C.; Zhang, G.; Li, F.; Tao, E. Rifampicin protects PC12 cells against MPP+-induced apoptosis and inhibits the expression of an alpha-Synuclein multimer. Brain Res. 2007, 1139, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acuña, L.; Hamadat, S.; Corbalán, N.S.; González-Lizárraga, F.; dos-Santos-Pereira, M.; Rocca, J.; Sepúlveda Díaz, J.; Del-Bel, E.; Papy-García, D.; Chehín, R.N.; et al. Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neurodegeneration Induced In Vitro by α-Synuclein Fibrillary Aggregates. Cells 2019, 8, 776. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080776
Acuña L, Hamadat S, Corbalán NS, González-Lizárraga F, dos-Santos-Pereira M, Rocca J, Sepúlveda Díaz J, Del-Bel E, Papy-García D, Chehín RN, et al. Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neurodegeneration Induced In Vitro by α-Synuclein Fibrillary Aggregates. Cells. 2019; 8(8):776. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080776
Chicago/Turabian StyleAcuña, Leonardo, Sabah Hamadat, Natalia S. Corbalán, Florencia González-Lizárraga, Mauricio dos-Santos-Pereira, Jérémy Rocca, Julia Sepúlveda Díaz, Elaine Del-Bel, Dulce Papy-García, Rosana N. Chehín, and et al. 2019. "Rifampicin and Its Derivative Rifampicin Quinone Reduce Microglial Inflammatory Responses and Neurodegeneration Induced In Vitro by α-Synuclein Fibrillary Aggregates" Cells 8, no. 8: 776. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080776