Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus


Abstract
:1. Introduction
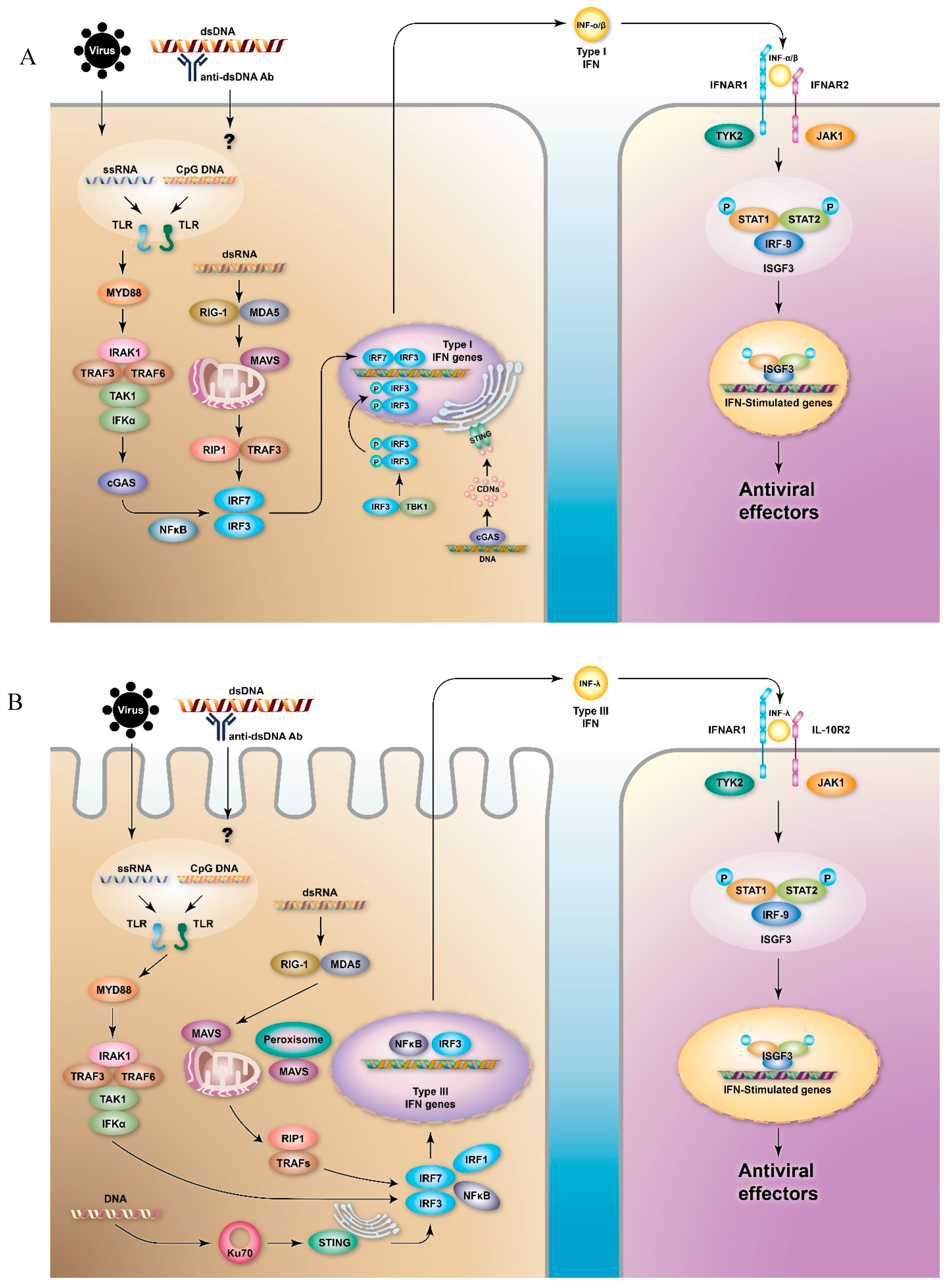
2. IFNs and IFN Signaling Pathways
2.1. Type I IFNs
2.2. Type III IFNs
3. Immunoregulatory Function of Type I and Type III IFNs
4. Type I and Type III IFNs in Autoimmunity
5. Type I IFNs in SLE
6. Type III IFNs in SLE
7. Targeting Type I IFN and IFN Signaling Pathways in SLE
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feldman, C.H.; Hiraki, L.T.; Liu, J.; Fischer, M.A.; Solomon, D.H.; Alarcon, G.S.; Winkelmayer, W.C.; Costenbader, K.H. Epidemiology and sociodemographics of systemic lupus erythematosus and lupus nephritis among US adults with Medicaid coverage, 2000–2004. Arthritis Rheum. 2013, 65, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.G.; Stein, C.M. Systemic lupus erythematosus in Zimbabwe. Ann. Rheum. Dis. 1986, 45, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Rees, F.; Doherty, M.; Grainge, M.J.; Lanyon, P.; Zhang, W. The worldwide incidence and prevalence of systemic lupus erythematosus: A systematic review of epidemiological studies. Rheumatology (Oxford) 2017, 56, 1945–1961. [Google Scholar] [CrossRef] [PubMed]
- Gorji, A.E.; Roudbari, Z.; Alizadeh, A.; Sadeghi, B. Investigation of systemic lupus erythematosus (SLE) with integrating transcriptomics and genome wide association information. Gene 2019, 706, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Tsao, B.P. Updates in Lupus Genetics. Curr. Rheumatol. Rep. 2017, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Moser, K.L.; Kelly, J.A.; Lessard, C.J.; Harley, J.B. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009, 10, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Davidson, A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat. Med. 2012, 18, 871–882. [Google Scholar] [CrossRef]
- Li, Q.Z.; Zhou, J.; Lian, Y.; Zhang, B.; Branch, V.K.; Carr-Johnson, F.; Karp, D.R.; Mohan, C.; Wakeland, E.K.; Olsen, N.J. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin. Exp. Immunol. 2010, 159, 281–291. [Google Scholar] [CrossRef]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Touma, Z.; Gladman, D.D. Current and future therapies for SLE: Obstacles and recommendations for the development of novel treatments. Lupus Sci. Med. 2017, 4, e000239. [Google Scholar] [CrossRef]
- Green, D.S.; Young, H.A.; Valencia, J.C. Current prospects of type II interferon gamma signaling and autoimmunity. J. Biol. Chem. 2017, 292, 13925–13933. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Mantei, N.; Schwarzstein, M.; Nagata, S.; Muramatsu, M.; Weissmann, C. Human leukocyte and fibroblast interferons are structurally related. Nature 1980, 285, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Elkon, K.B.; Fitzgerald, K.A. Importance of Nucleic Acid Recognition in Inflammation and Autoimmunity. Annu. Rev. Med. 2016, 67, 323–336. [Google Scholar] [CrossRef]
- Mackern-Oberti, J.P.; Llanos, C.; Vega, F.; Salazar-Onfray, F.; Riedel, C.A.; Bueno, S.M.; Kalergis, A.M. Role of dendritic cells in the initiation, progress and modulation of systemic autoimmune diseases. Autoimmun. Rev. 2015, 14, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Gilliet, M.; Cao, W.; Liu, Y.J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef]
- Smith, N.; Vidalain, P.O.; Nisole, S.; Herbeuval, J.P. An efficient method for gene silencing in human primary plasmacytoid dendritic cells: Silencing of the TLR7/IRF-7 pathway as a proof of concept. Sci. Rep. 2016, 6, 29891. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef]
- McCaffary, D. STING signalling: An emerging common pathway in autoimmunity and cancer. Immunopharmacol. Immunotoxicol. 2017, 39, 253–258. [Google Scholar] [CrossRef]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef]
- Negrotto, S.C.J.D.G.; Lapponi, M.J.; Etulain, J.; Rivadeneyra, L.; Pozner, R.G.; Gomez, R.M.; Schattner, M. Expression and functionality of type I interferon receptor in the megakaryocytic lineage. J. Thromb. Haemost. 2011, 9, 2477–2485. [Google Scholar] [CrossRef]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [Green Version]
- van Boxel-Dezaire, A.H.; Rani, M.R.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Holzl, E.; Rosebrock, F.; Muller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, K.; Witte, E.; Proesch, S.; Schulze-Tanzil, G.; Nasilowska, K.; Thilo, J.; Asadullah, K.; Sterry, W.; Volk, H.D.; et al. Maturing dendritic cells are an important source of IL-29 and IL-20 that may cooperatively increase the innate immunity of keratinocytes. J. Leukoc. Biol. 2008, 83, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Dai, J.; Deng, J.; Sheikh, F.; Natalia, M.; Shih, T.; Lewis-Antes, A.; Amrute, S.B.; Garrigues, U.; Doyle, S.; et al. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J. Immunol. 2012, 189, 2735–2745. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Mahlakoiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Jewell, N.A.; Cline, T.; Mertz, S.E.; Smirnov, S.V.; Flano, E.; Schindler, C.; Grieves, J.L.; Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J. Virol. 2010, 84, 11515–11522. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; Iversen, M.B.; Bartholdy, C.; Staeheli, P.; Hartmann, R.; Jensen, U.B.; Dagnaes-Hansen, F.; Thomsen, A.R.; Chen, Z.; Haugen, H.; et al. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J. Immunol. 2008, 180, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING is an essential mediator of the Ku70-mediated production of IFN-lambda1 in response to exogenous DNA. Sci. Signal 2017, 10. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [PubMed]
- Bolen, C.R.; Ding, S.; Robek, M.D.; Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef]
- Lin, J.D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct Roles of Type I and Type III Interferons in Intestinal Immunity to Homologous and Heterologous Rotavirus Infections. PLoS Pathog. 2016, 12, e1005600. [Google Scholar] [CrossRef]
- Thomson, S.J.; Goh, F.G.; Banks, H.; Krausgruber, T.; Kotenko, S.V.; Foxwell, B.M.; Udalova, I.A. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11564–11569. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, T.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Involvement of NK Cells in IL-28B-Mediated Immunity against Influenza Virus Infection. J. Immunol. 2017, 199, 1012–1020. [Google Scholar] [CrossRef]
- Megjugorac, N.J.; Gallagher, G.E.; Gallagher, G. Modulation of human plasmacytoid DC function by IFN-lambda1 (IL-29). J. Leukoc. Biol. 2009, 86, 1359–1363. [Google Scholar] [CrossRef]
- Koltsida, O.; Hausding, M.; Stavropoulos, A.; Koch, S.; Tzelepis, G.; Ubel, C.; Kotenko, S.V.; Sideras, P.; Lehr, H.A.; Tepe, M.; et al. IL-28A (IFN-lambda2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 2011, 3, 348–361. [Google Scholar] [CrossRef]
- Paquette, R.L.; Hsu, N.C.; Kiertscher, S.M.; Park, A.N.; Tran, L.; Roth, M.D.; Glaspy, J.A. Interferon-alpha and granulocyte-macrophage colony-stimulating factor differentiate peripheral blood monocytes into potent antigen-presenting cells. J. Leukoc. Biol. 1998, 64, 358–367. [Google Scholar] [CrossRef]
- Santini, S.M.; Lapenta, C.; Logozzi, M.; Parlato, S.; Spada, M.; Di Pucchio, T.; Belardelli, F. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J. Exp. Med. 2000, 191, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.P.; Wearsch, P.A.; Canaday, D.H.; Meyerson, H.J.; Liu, Y.C.; Wang, Y.; Boom, W.H.; Harding, C.V. Type I IFN drives a distinctive dendritic cell maturation phenotype that allows continued class II MHC synthesis and antigen processing. J. Immunol. 2012, 188, 3116–3126. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, S.; Nicola, S.; Riva, A.; Biasin, M.; Clerici, M.; Villa, M.L. Functional repertoire of dendritic cells generated in granulocyte macrophage-colony stimulating factor and interferon-alpha. J. Leukoc. Biol. 2004, 75, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Rouzaut, A.; Garasa, S.; Teijeira, A.; Gonzalez, I.; Martinez-Forero, I.; Suarez, N.; Larrea, E.; Alfaro, C.; Palazon, A.; Dubrot, J.; et al. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-alpha. Eur. J. Immunol. 2010, 40, 3054–3063. [Google Scholar] [CrossRef] [PubMed]
- Padovan, E.; Spagnoli, G.C.; Ferrantini, M.; Heberer, M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J. Leukoc. Biol. 2002, 71, 669–676. [Google Scholar] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Tanabe, Y.; Nishibori, T.; Su, L.; Arduini, R.M.; Baker, D.P.; David, M. Cutting edge: Role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J. Immunol. 2005, 174, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, R.; Lee, C.K.; Schindler, C.; Levy, D.E. Stat1 and Stat2 but not Stat3 arbitrate contradictory growth signals elicited by alpha/beta interferon in T lymphocytes. Mol. Cell Biol. 2005, 25, 5456–5465. [Google Scholar] [CrossRef]
- Van De Wiele, C.J.; Marino, J.H.; Whetsell, M.E.; Vo, S.S.; Masengale, R.M.; Teague, T.K. Loss of interferon-induced Stat1 phosphorylation in activated T cells. J. Interferon Cytokine Res. 2004, 24, 169–178. [Google Scholar] [CrossRef]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Le Bon, A.; Thompson, C.; Kamphuis, E.; Durand, V.; Rossmann, C.; Kalinke, U.; Tough, D.F. Cutting edge: Enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 2006, 176, 2074–2078. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Dong, C.; Cooper, M.D. Impairment of T and B cell development by treatment with a type I interferon. J. Exp. Med. 1998, 187, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Fallet, B.; Narr, K.; Ertuna, Y.I.; Remy, M.; Sommerstein, R.; Cornille, K.; Kreutzfeldt, M.; Page, N.; Zimmer, G.; Geier, F.; et al. Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Moseman, E.A.; Wu, T.; de la Torre, J.C.; Schwartzberg, P.L.; McGavern, D.B. Type I interferon suppresses virus-specific B cell responses by modulating CD8(+) T cell differentiation. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef]
- Liu, M.; Guo, Q.; Wu, C.; Sterlin, D.; Goswami, S.; Zhang, Y.; Li, T.; Bao, C.; Shen, N.; Fu, Q.; et al. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cell Mol. Immunol. 2019, 16, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Morrow, M.P.; Pankhong, P.; Laddy, D.J.; Schoenly, K.A.; Yan, J.; Cisper, N.; Weiner, D.B. Comparative ability of IL-12 and IL-28B to regulate Treg populations and enhance adaptive cellular immunity. Blood 2009, 113, 5868–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Megjugorac, N.J.; Gallagher, G.E.; Yu, R.Y.; Gallagher, G. IFN-lambda1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood 2009, 113, 5829–5838. [Google Scholar] [CrossRef] [PubMed]
- Jordan, W.J.; Eskdale, J.; Srinivas, S.; Pekarek, V.; Kelner, D.; Rodia, M.; Gallagher, G. Human interferon lambda-1 (IFN-lambda1/IL-29) modulates the Th1/Th2 response. Genes Immun. 2007, 8, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Santer, D.M.; O’Shea, D.; Barakat, K.; Syedbasha, M.; Vollmer, M.; Baluch, A.; Bhat, R.; Groenendyk, J.; Joyce, M.A.; et al. IL-28B is a key regulator of B- and T-cell vaccine responses against influenza. PLoS Pathog. 2014, 10, e1004556. [Google Scholar] [CrossRef]
- de Groen, R.A.; Groothuismink, Z.M.; Liu, B.S.; Boonstra, A. IFN-lambda is able to augment TLR-mediated activation and subsequent function of primary human B cells. J. Leukoc. Biol. 2015, 98, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; Kind, B.; Reijns, M.A.; Berndt, N.; Martinez-Bueno, M.; Wolf, C.; Tungler, V.; Chara, O.; Lee, Y.A.; Hubner, N.; et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Investig. 2015, 125, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskaite, Z.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B.; et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 2016, 35, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Campbell, A.M.; Chan, J.; Schattgen, S.A.; Orlowski, G.M.; Nayar, R.; Huyler, A.H.; Nundel, K.; Mohan, C.; Berg, L.J.; et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc. Natl. Acad. Sci. USA 2015, 112, E710–E717. [Google Scholar] [CrossRef] [PubMed]
- Jeremiah, N.; Neven, B.; Gentili, M.; Callebaut, I.; Maschalidi, S.; Stolzenberg, M.C.; Goudin, N.; Fremond, M.L.; Nitschke, P.; Molina, T.J.; et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Investig. 2014, 124, 5516–5520. [Google Scholar] [CrossRef] [PubMed]
- Orcesi, S.; La Piana, R.; Fazzi, E. Aicardi-Goutieres syndrome. Br. Med. Bull. 2009, 89, 183–201. [Google Scholar] [CrossRef]
- Ablasser, A.; Hemmerling, I.; Schmid-Burgk, J.L.; Behrendt, R.; Roers, A.; Hornung, V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014, 192, 5993–5997. [Google Scholar] [CrossRef]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep. 2016, 16, 1492–1501. [Google Scholar] [CrossRef] [Green Version]
- Sjostrand, M.; Johansson, A.; Aqrawi, L.; Olsson, T.; Wahren-Herlenius, M.; Espinosa, A. The Expression of BAFF Is Controlled by IRF Transcription Factors. J. Immunol. 2016, 196, 91–96. [Google Scholar] [CrossRef]
- Maria, N.I.; Steenwijk, E.C.; AS, I.J.; van Helden-Meeuwsen, C.G.; Vogelsang, P.; Beumer, W.; Brkic, Z.; van Daele, P.L.; van Hagen, P.M.; van der Spek, P.J.; et al. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjogren’s syndrome. Ann. Rheum. Dis. 2017, 76, 721–730. [Google Scholar] [CrossRef]
- Rodriguez-Carrio, J.; Lopez, P.; Suarez, A. Type I IFNs as biomarkers in rheumatoid arthritis: Towards disease profiling and personalized medicine. Clin. Sci. (Lond.) 2015, 128, 449–464. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Q.; Sun, H.; Li, M.; Zhang, Y.; La Cava, A. Serum IFN-lambda1 is abnormally elevated in rheumatoid arthritis patients. Autoimmunity 2013, 46, 40–43. [Google Scholar] [CrossRef]
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. IFN-lambda resolves inflammation via suppression of neutrophil infiltration and IL-1beta production. J. Exp. Med. 2015, 212, 845–853. [Google Scholar] [CrossRef]
- Broggi, A.; Tan, Y.; Granucci, F.; Zanoni, I. IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol. 2017, 18, 1084–1093. [Google Scholar] [CrossRef]
- Wolk, K.; Witte, K.; Witte, E.; Raftery, M.; Kokolakis, G.; Philipp, S.; Schonrich, G.; Warszawska, K.; Kirsch, S.; Prosch, S.; et al. IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Witte, E.; Kokolakis, G.; Witte, K.; Warszawska, K.; Friedrich, M.; Christou, D.; Kirsch, S.; Sterry, W.; Volk, H.D.; Sabat, R.; et al. Interleukin-29 induces epithelial production of CXCR3A ligands and T-cell infiltration. J. Mol. Med. (Berl.) 2016, 94, 391–400. [Google Scholar] [CrossRef]
- Steinberg, A.D.; Baron, S.; Talal, N. The pathogenesis of autoimmunity in New Zealand mice, I. Induction of antinucleic acid antibodies by polyinosinic-polycytidylic acid. Proc. Natl. Acad. Sci. USA 1969, 63, 1102–1107. [Google Scholar] [CrossRef]
- Hooks, J.J.; Moutsopoulos, H.M.; Geis, S.A.; Stahl, N.I.; Decker, J.L.; Notkins, A.L. Immune interferon in the circulation of patients with autoimmune disease. N. Engl. J. Med. 1979, 301, 5–8. [Google Scholar] [CrossRef]
- Dumoulin, F.L.; Leifeld, L.; Sauerbruch, T.; Spengler, U. Autoimmunity induced by interferon-alpha therapy for chronic viral hepatitis. Biomed. Pharmacother. 1999, 53, 242–254. [Google Scholar] [CrossRef]
- Ronnblom, L.E.; Alm, G.V.; Oberg, K.E. Autoimmunity after alpha-interferon therapy for malignant carcinoid tumors. Ann. Intern. Med. 1991, 115, 178–183. [Google Scholar] [CrossRef]
- Sigurdsson, S.; Nordmark, G.; Garnier, S.; Grundberg, E.; Kwan, T.; Nilsson, O.; Eloranta, M.L.; Gunnarsson, I.; Svenungsson, E.; Sturfelt, G.; et al. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum. Mol. Genet. 2008, 17, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- Abelson, A.K.; Delgado-Vega, A.M.; Kozyrev, S.V.; Sanchez, E.; Velazquez-Cruz, R.; Eriksson, N.; Wojcik, J.; Linga Reddy, M.V.; Lima, G.; D’Alfonso, S.; et al. STAT4 associates with systemic lupus erythematosus through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk. Ann. Rheum. Dis. 2009, 68, 1746–1753. [Google Scholar] [CrossRef]
- Harley, I.T.; Kaufman, K.M.; Langefeld, C.D.; Harley, J.B.; Kelly, J.A. Genetic susceptibility to SLE: New insights from fine mapping and genome-wide association studies. Nat. Rev. Genet. 2009, 10, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J. Type I interferonopathies: A novel set of inborn errors of immunity. Ann. N. Y. Acad. Sci. 2011, 1238, 91–98. [Google Scholar] [CrossRef]
- Gonzalez-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef]
- Eloranta, M.L.; Alm, G.V.; Ronnblom, L. Disease mechanisms in rheumatology--tools and pathways: Plasmacytoid dendritic cells and their role in autoimmune rheumatic diseases. Arthritis Rheum. 2013, 65, 853–863. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar] [CrossRef]
- Le Buanec, H.; Gougeon, M.L.; Mathian, A.; Lebon, P.; Dupont, J.M.; Peltre, G.; Hemon, P.; Schmid, M.; Bizzini, B.; Kunding, T.; et al. IFN-alpha and CD46 stimulation are associated with active lupus and skew natural T regulatory cell differentiation to type 1 regulatory T (Tr1) cells. Proc. Natl. Acad. Sci. USA 2011, 108, 18995–19000. [Google Scholar] [CrossRef]
- Moschen, A.R.; Geiger, S.; Krehan, I.; Kaser, A.; Tilg, H. Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology 2008, 213, 779–787. [Google Scholar] [CrossRef]
- Liu, J.; Berthier, C.C.; Kahlenberg, J.M. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol. 2017, 69, 1840–1849. [Google Scholar] [CrossRef]
- Interferon beta-1b in the treatment of multiple sclerosis: Final outcome of the randomized controlled trial. The IFNB Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 1995, 45, 1277–1285. [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Forster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Wu, Q.; Yang, P.; Luo, B.; Liu, S.; Li, J.; A, L.M.; Sanz, I.; Chatham, W.W.; Hsu, H.C.; et al. Cutting Edge: Intracellular IFN-beta and Distinct Type I IFN Expression Patterns in Circulating Systemic Lupus Erythematosus B Cells. J. Immunol. 2018, 201, 2203–2208. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, D.W.; Nardelli, B.; Tsareva, T.; Mather, D.; Feng, P.; Semenuk, M.; Taylor, K.; Buergin, M.; Chinchilla, D.; Roshke, V.; et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 2001, 276, 39765–39771. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, B.; Zaritskaya, L.; Semenuk, M.; Cho, Y.H.; LaFleur, D.W.; Shah, D.; Ullrich, S.; Girolomoni, G.; Albanesi, C.; Moore, P.A. Regulatory effect of IFN-kappa, a novel type I IFN, on cytokine production by cells of the innate immune system. J. Immunol. 2002, 169, 4822–4830. [Google Scholar] [CrossRef] [PubMed]
- Harley, I.T.; Niewold, T.B.; Stormont, R.M.; Kaufman, K.M.; Glenn, S.B.; Franek, B.S.; Kelly, J.A.; Kilpatrick, J.R.; Hutchings, D.; Divers, J.; et al. The role of genetic variation near interferon-kappa in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Stannard, J.N.; Reed, T.J.; Myers, E.; Lowe, L.; Sarkar, M.K.; Xing, X.; Gudjonsson, J.E.; Kahlenberg, J.M. Lupus Skin Is Primed for IL-6 Inflammatory Responses through a Keratinocyte-Mediated Autocrine Type I Interferon Loop. J. Investig. Dermatol. 2017, 137, 115–122. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, Q.; Lourenco, E.; Sun, H.; Zhang, Y. Interferon-lambda1 induces peripheral blood mononuclear cell-derived chemokines secretion in patients with systemic lupus erythematosus: Its correlation with disease activity. Arthritis Res. Ther. 2011, 13, R88. [Google Scholar] [CrossRef]
- Oke, V.; Brauner, S.; Larsson, A.; Gustafsson, J.; Zickert, A.; Gunnarsson, I.; Svenungsson, E. IFN-lambda1 with Th17 axis cytokines and IFN-alpha define different subsets in systemic lupus erythematosus (SLE). Arthritis Res. Ther. 2017, 19, 139. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wang, C.M.; Chen, T.D.; Jan Wu, Y.J.; Lin, J.C.; Lu, L.Y.; Wu, J. Interferon-lambda3/4 genetic variants and interferon-lambda3 serum levels are biomarkers of lupus nephritis and disease activity in Taiwanese. Arthritis Res. Ther. 2018, 20, 193. [Google Scholar] [CrossRef]
- Zickert, A.; Oke, V.; Parodis, I.; Svenungsson, E.; Sundstrom, Y.; Gunnarsson, I. Interferon (IFN)-lambda is a potential mediator in lupus nephritis. Lupus Sci. Med. 2016, 3, e000170. [Google Scholar] [CrossRef]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjall, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef]
- Amezcua-Guerra, L.M.; Marquez-Velasco, R.; Chavez-Rueda, A.K.; Castillo-Martinez, D.; Masso, F.; Paez, A.; Colin-Fuentes, J.; Bojalil, R. Type III Interferons in Systemic Lupus Erythematosus: Association Between Interferon lambda3, Disease Activity, and Anti-Ro/SSA Antibodies. J. Clin. Rheumatol. 2017, 23, 368–375. [Google Scholar] [CrossRef]
- Kalunian, K.C.; Merrill, J.T.; Maciuca, R.; McBride, J.M.; Townsend, M.J.; Wei, X.; Davis, J.C., Jr.; Kennedy, W.P. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann. Rheum. Dis. 2016, 75, 196–202. [Google Scholar] [CrossRef]
- Khamashta, M.; Merrill, J.T.; Werth, V.P.; Furie, R.; Kalunian, K.; Illei, G.G.; Drappa, J.; Wang, L.; Greth, W.; CD1067 Study Investigators. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: A randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 1909–1916. [Google Scholar] [CrossRef]
- Tcherepanova, I.; Curtis, M.; Sale, M.; Miesowicz, F.; Nicolette, C. SAT0193 Results of a randomized placebo controlled phase ia study of AGS-009, a humanized anti-interferon-α monoclonal antibody in subjects with systemic lupus erythematosus. Ann. Rheum. Dis. 2014, 71, 533–537. [Google Scholar] [CrossRef]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef]
- Lauwerys, B.R.; Hachulla, E.; Spertini, F.; Lazaro, E.; Jorgensen, C.; Mariette, X.; Haelterman, E.; Grouard-Vogel, G.; Fanget, B.; Dhellin, O.; et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon alpha-kinoid. Arthritis Rheum. 2013, 65, 447–456. [Google Scholar] [CrossRef]
- Wallace, D.J.; Furie, R.A.; Tanaka, Y.; Kalunian, K.C.; Mosca, M.; Petri, M.A.; Dorner, T.; Cardiel, M.H.; Bruce, I.N.; Gomez, E.; et al. Baricitinib for systemic lupus erythematosus: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 222–231. [Google Scholar] [CrossRef]
- Chan, E.S.; Herlitz, L.C.; Jabbari, A. Ruxolitinib Attenuates Cutaneous Lupus Development in a Mouse Lupus Model. J. Investig. Dermatol. 2015, 135, 1912–1915. [Google Scholar] [CrossRef] [Green Version]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis. Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef]
- Merrill, J.T.; Wallace, D.J.; Petri, M.; Kirou, K.A.; Yao, Y.; White, W.I.; Robbie, G.; Levin, R.; Berney, S.M.; Chindalore, V.; et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: A phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis. 2011, 70, 1905–1913. [Google Scholar] [CrossRef]
- Mathian, A.; Amoura, Z.; Adam, E.; Colaone, F.; Hoekman, M.F.; Dhellin, O.; Vandepapeliere, P.; Haroche, J.; Piette, J.C.; Lebon, P.; et al. Active immunisation of human interferon alpha transgenic mice with a human interferon alpha Kinoid induces antibodies that neutralise interferon alpha in sera from patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2011, 70, 1138–1143. [Google Scholar] [CrossRef]
- Zagury, D.; Le Buanec, H.; Mathian, A.; Larcier, P.; Burnett, R.; Amoura, Z.; Emilie, D.; Peltre, G.; Bensussan, A.; Bizzini, B.; et al. IFNalpha kinoid vaccine-induced neutralizing antibodies prevent clinical manifestations in a lupus flare murine model. Proc. Natl. Acad. Sci. USA 2009, 106, 5294–5299. [Google Scholar] [CrossRef]
- Medina-Rosas, J.; Al-Rayes, H.; Moustafa, A.T.; Touma, Z. Recent advances in the biologic therapy of lupus: The 10 most important areas to look for common pitfalls in clinical trials. Expert Opin. Biol. Ther. 2016, 16, 1225–1238. [Google Scholar] [CrossRef]
- Rodriguez-Pinto, I.; Espinosa, G.; Cervera, R. The problems and pitfalls in systemic lupus erythematosus drug discovery. Expert Opin. Drug Discov. 2016, 11, 525–527. [Google Scholar] [CrossRef] [Green Version]
- Wrobleski, S.T.; Moslin, R.; Lin, S.; Zhang, Y.; Spergel, S.; Kempson, J.; Tokarski, J.S.; Strnad, J.; Zupa-Fernandez, A.; Cheng, L.; et al. Highly Selective Inhibition of Tyrosine Kinase 2 (TYK2) for the Treatment of Autoimmune Diseases: Discovery of the Allosteric Inhibitor BMS-986165. J. Med. Chem. 2019. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Therapy | Mechanism of Action | Current Development Stage | Ref. |
|---|---|---|---|
| Rontalizumab | Humanized IgG1 mAb against IFN-α | Phase II, completed | [115] |
| Sifalimumab | Fully human IgG1 against IFN-α | Phase IIb, completed | [116] |
| AGS-009 | Humanized IgG4 mAb against IFN-α | Phase I, completed | [117] |
| Anifrolumab | Fully human IgG1κ mAb against IFNAR1 | Phase II (MUSE trial), completed Phase III (TULIP 1 and 2), completed | [118] NCT02446899 NCT02446912 |
| IFN-α kinoid (IFN-K) | Therapeutic vaccine of IFN-α2b | Phase I/II, completed | [119] |
| Baricitinib | JAK1/JAK2 inhibitor | Phase II, completed Phase III, recruiting | [120] NCT03616964 NCT03616912 |
| Tofacitinib | JAK1/JAK2/JAK3 inhibitor | Phase I, completed Phase I/II, recruiting | NCT02535689 NCT03288324 |
| Ruxolitinib | JAK1/JAK2 inhibitor | Preclinical | [121] |
| CC-930 | JAK1/JAK2/JAK3 inhibitor | Phase II, terminated (discoid lupus) | NCT01466725 |
| GSK2586184 | JAK1 inhibitor | Phase I, completed Phase II, terminated | NCT01687309 NCT01777256 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chyuan, I.-T.; Tzeng, H.-T.; Chen, J.-Y. Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus. Cells 2019, 8, 963. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8090963
Chyuan I-T, Tzeng H-T, Chen J-Y. Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus. Cells. 2019; 8(9):963. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8090963
Chicago/Turabian StyleChyuan, I-Tsu, Hong-Tai Tzeng, and Ji-Yih Chen. 2019. "Signaling Pathways of Type I and Type III Interferons and Targeted Therapies in Systemic Lupus Erythematosus" Cells 8, no. 9: 963. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8090963