Human Autopsy-Derived Scalp Fibroblast Biobanking for Age-Related Neurodegenerative Disease Research

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Clinical and Neuropathological Diagnosis
2.3. Features of the Donors
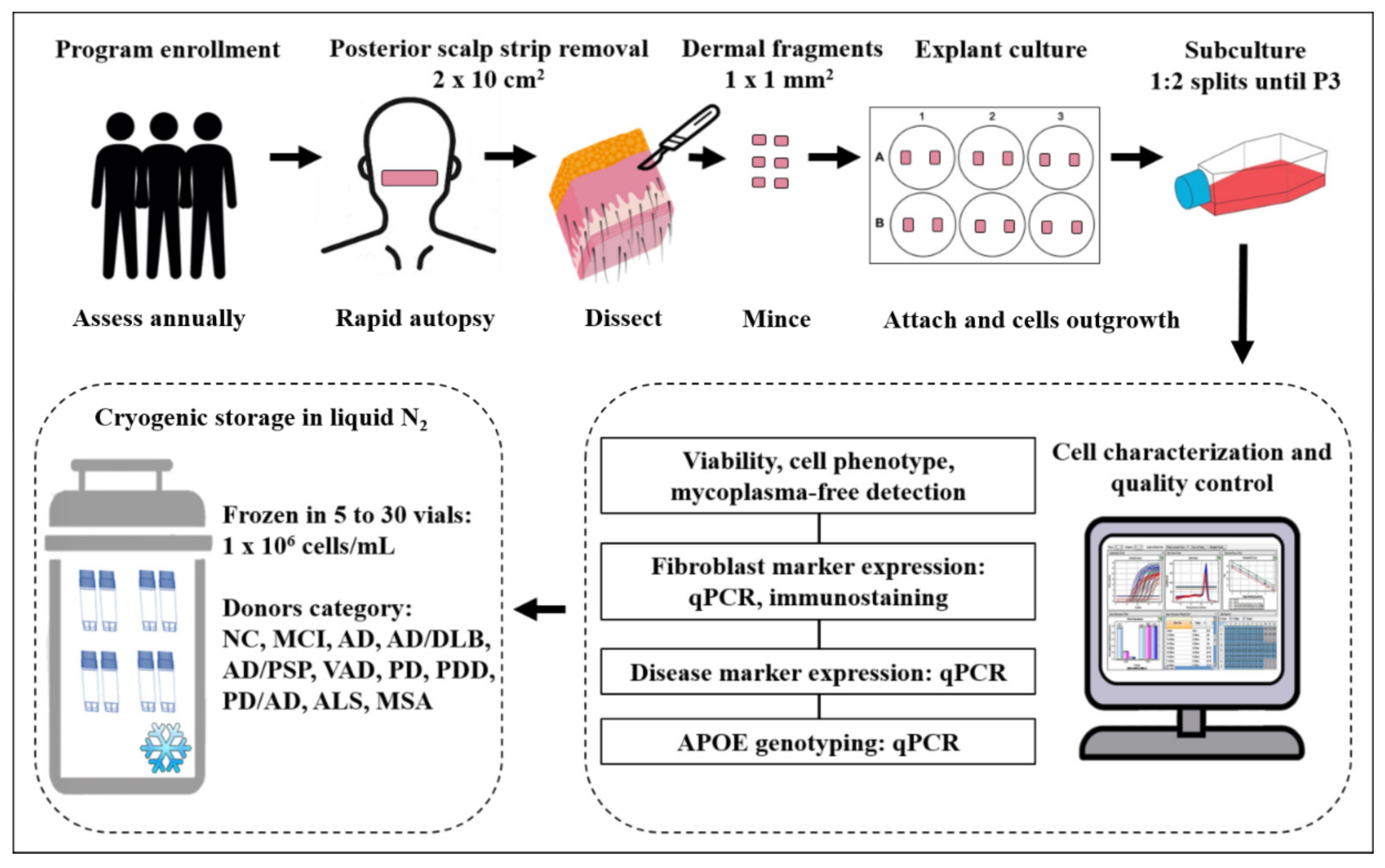
2.4. Tissue Processing and Culture
2.5. Passage and Cryoprotection
2.6. Recovery Rate of Cryoprotected Fibroblasts
2.7. Immunohistochemistry, Immunofluorescence, and Microscopy
2.8. RNA Isolation and cDNA Synthesis
2.9. Quantitative Real-Time Polymerase Chain Reaction
2.10. DNA Extraction and Apolipoprotein E (APOE) Genotyping
2.11. Generation and Characterization of Human Pluripotent Stem Cells
2.12. Statistical Analysis
3. Results
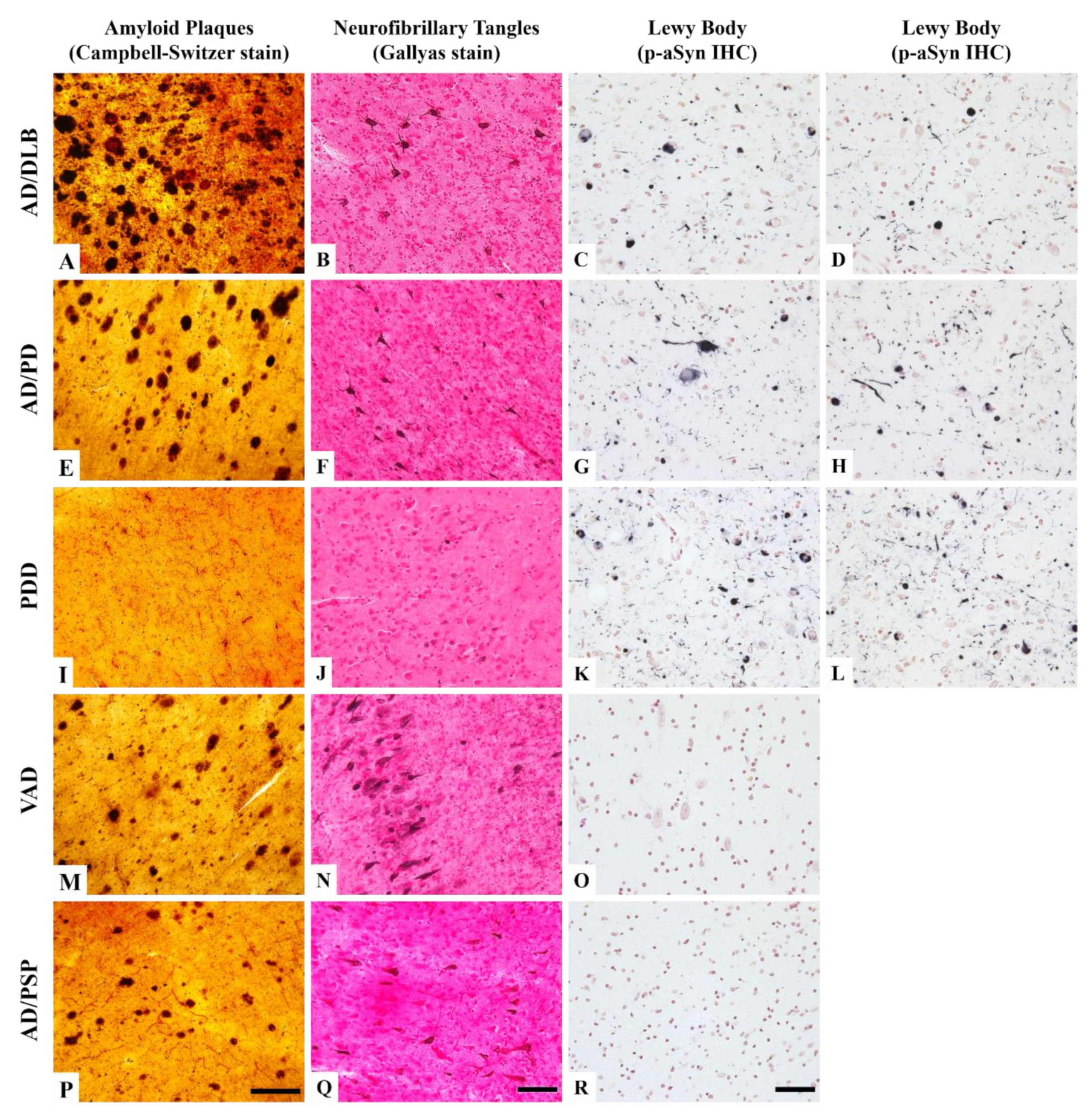
3.1. Neuropathological Features of the Donors
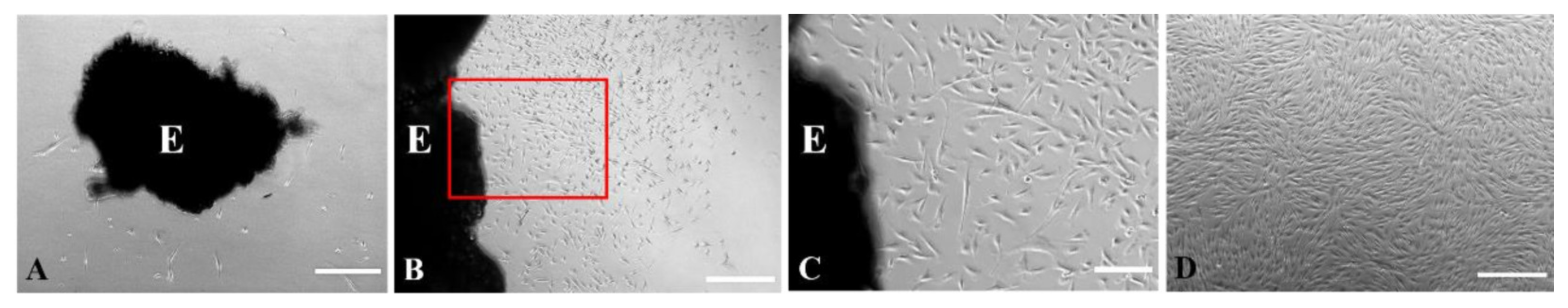
3.2. The Explant and Cell Cultures
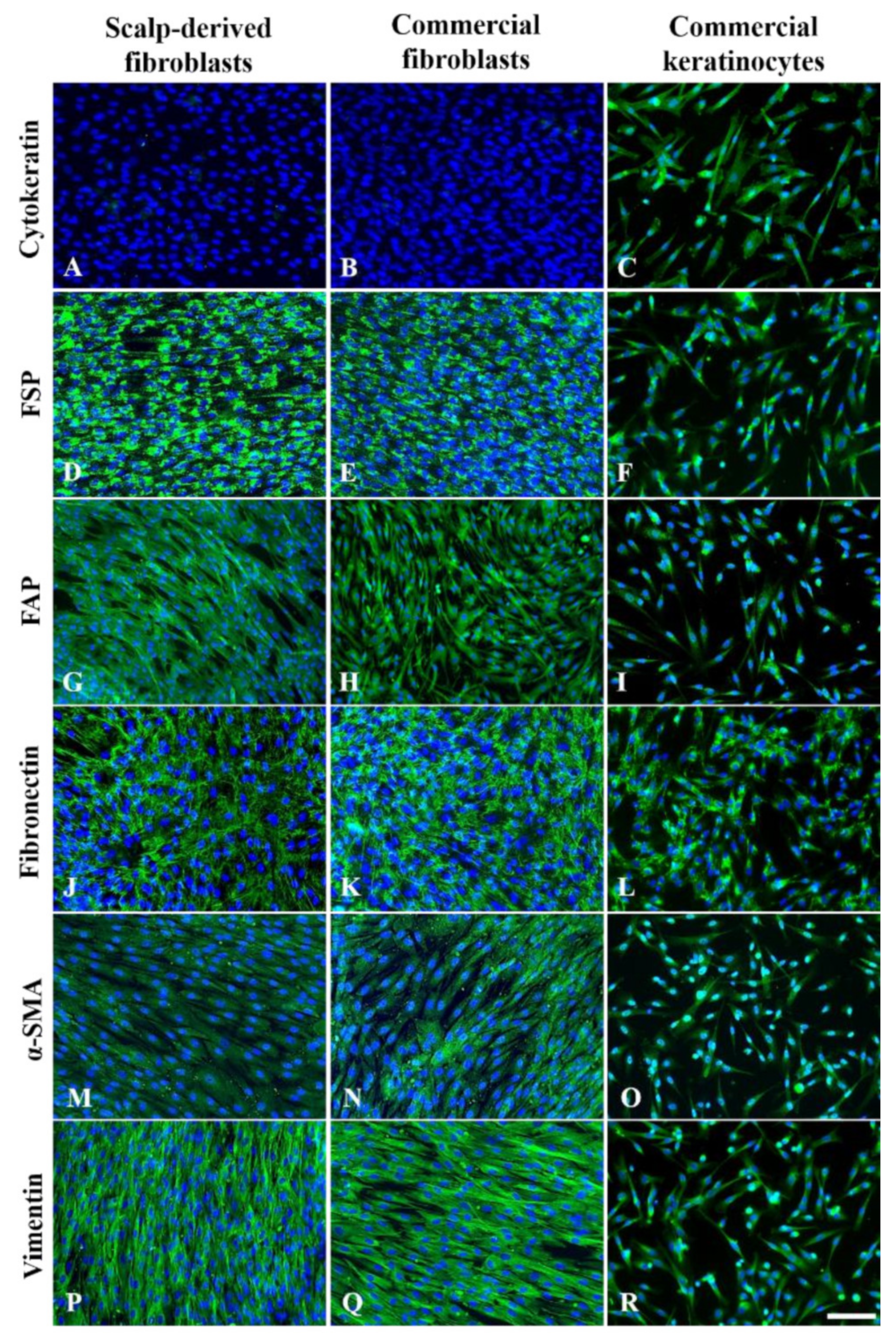
3.3. Immunofluorescence Characterization of P3 Fibroblasts
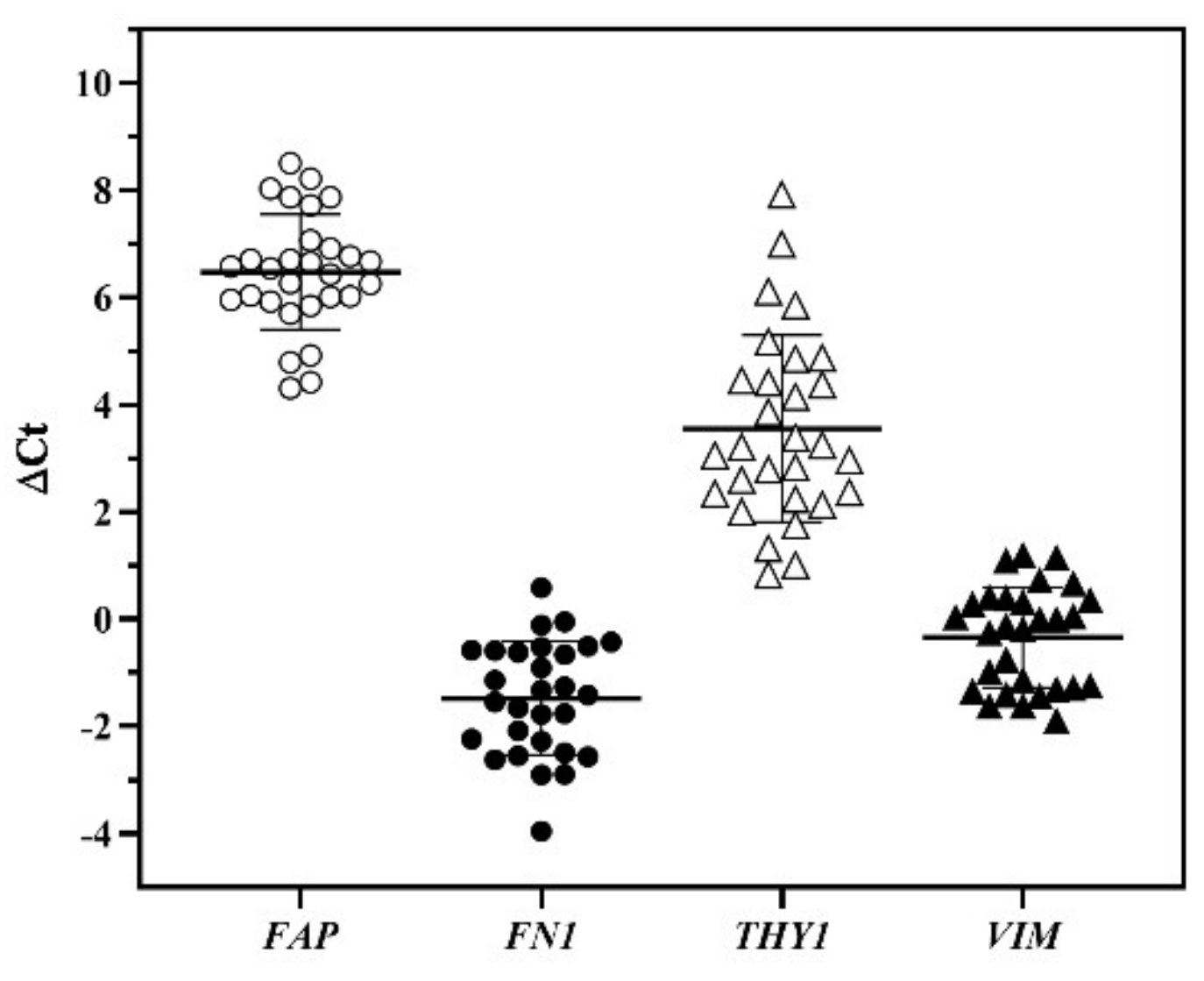
3.4. Characterization of Fibroblast Cultures by qPCR Analysis
3.5. Fibroblast Gene Expression and Culture Time Course
3.6. Quality Control for Cell Banking
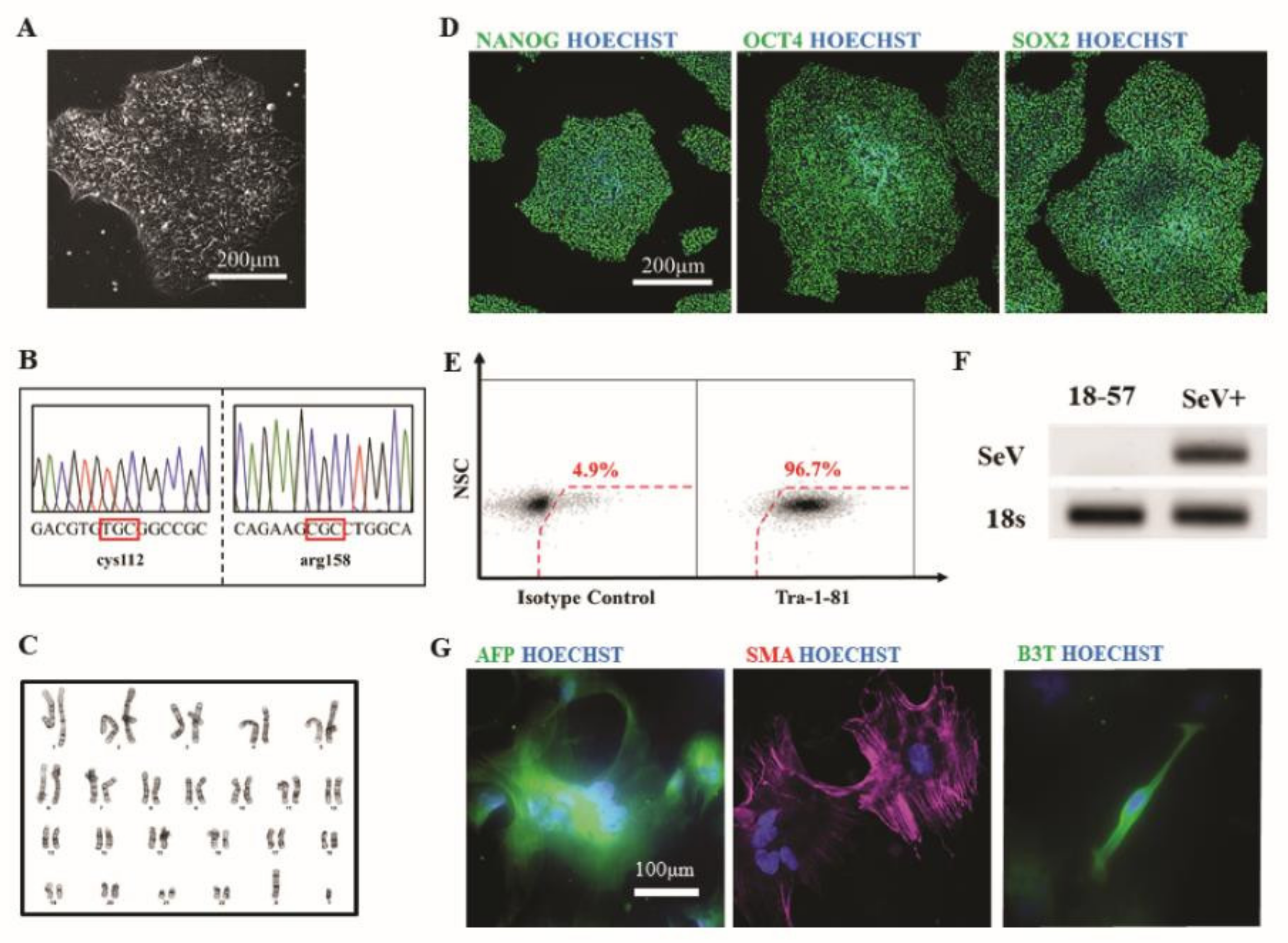
3.7. Generation of hiPSCs from Patient Fibroblasts
4. Discussion
4.1. Diverse Patient Phenotypes
4.2. The Features of the Culturing Procedure
4.3. The Features of Postmortem Scalp Fibroblasts
4.4. Future of the Human-Derived Fibroblast Banking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Puzzo, D.; Gulisano, W.; Palmeri, A.; Arancio, O. Rodent models for Alzheimer’s disease drug discovery. Expert Opin Drug Discov. 2015, 10, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.; Williams, M. Preclinical Models of Alzheimer’s Disease: Relevance and Translational Validity. Curr. Protoc. Pharm. 2019, 84, e57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ransohoff, R.M. All (animal) models (of neurodegeneration) are wrong. Are they also useful? J. Exp. Med. 2018, 215, 2955–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, C.M.; Hernández, D.; Wang, J.C.; Marsh, J.; Hewitt, A.W.; Hsu, S.; Norton, J.; Levitch, D.; Donahue, T.; Sigurdson, W.; et al. Human fibroblast and stem cell resource from the Dominantly Inherited Alzheimer Network. Alzheimers Res. 2018, 10, 69. [Google Scholar] [CrossRef]
- Simmnacher, K.; Lanfer, J.; Rizo, T.; Kaindl, J.; Winner, B. Modeling Cell-Cell Interactions in Parkinson’s Disease Using Human Stem Cell-Based Models. Front. Cell Neurosci. 2019, 13, 571. [Google Scholar] [CrossRef] [Green Version]
- Caiazzo, M.; Dell’Anno, M.T.; Dvoretskova, E.; Lazarevic, D.; Taverna, S.; Leo, D.; Sotnikova, T.D.; Menegon, A.; Roncaglia, P.; Colciago, G.; et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 2011, 476, 224–227. [Google Scholar] [CrossRef]
- Pfisterer, U.; Kirkeby, A.; Torper, O.; Wood, J.; Nelander, J.; Dufour, A.; Björklund, A.; Lindvall, O.; Jakobsson, J.; Parmar, M. Direct conversion of human fibroblasts to dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2011, 108, 10343–10348. [Google Scholar] [CrossRef] [Green Version]
- Ring, K.L.; Tong, L.M.; Balestra, M.E.; Javier, R.; Andrews-Zwilling, Y.; Li, G.; Walker, D.; Zhang, W.R.; Kreitzer, A.C.; Huang, Y. Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 2012, 11, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Miskinyte, G.; Devaraju, K.; Grønning Hansen, M.; Monni, E.; Tornero, D.; Woods, N.B.; Bengzon, J.; Ahlenius, H.; Lindvall, O.; Kokaia, Z. Direct conversion of human fibroblasts to functional excitatory cortical neurons integrating into human neural networks. Stem Cell Res. 2017, 8, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary skin fibroblasts as a model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Yakhine-Diop, S.M.S.; Niso-Santano, M.; Rodríguez-Arribas, M.; Gómez-Sánchez, R.; Martínez-Chacón, G.; Uribe-Carretero, E.; Navarro-García, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M.; et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2466–2481. [Google Scholar] [CrossRef]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Pereira, F.B.; Raimundo, N.; Oliveira, P.J. Mitochondrial remodeling in human skin fibroblasts from sporadic male Parkinson’s disease patients uncovers metabolic and mitochondrial bioenergetic defects. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165615. [Google Scholar] [CrossRef] [PubMed]
- Soininen, H.; Syrjänen, S.; Heinonen, O.; Neittaanmäki, H.; Miettinen, R.; Paljärvi, L.; Syrjänen, K.; Beyreuther, K.; Riekkinen, P. Amyloid beta-protein deposition in skin of patients with dementia. Lancet 1992, 339, 245. [Google Scholar] [CrossRef]
- Wen, G.Y.; Wisniewski, H.M.; Blondal, H.; Benedikz, E.; Frey, H.; Pirttila, T.; Rudelli, R.; Kim, K.S. Presence of non-fibrillar amyloid beta protein in skin biopsies of Alzheimer’s disease (AD), Down’s syndrome and non-AD normal persons. Acta Neuropathol. 1994, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Ponce, D.P.; Osorio-Fuentealba, C.; Behrens, M.I.; Quintanilla, R.A. Mitochondrial Bioenergetics Is Altered in Fibroblasts from Patients with Sporadic Alzheimer’s Disease. Front. Neurosci. 2017, 11, 553. [Google Scholar] [CrossRef]
- Zuev, V.A.; Dyatlova, A.S.; Lin’kova, N.S.; Kvetnoi, I.M.; Belushkina, N.N.; Pal’tsev, V.A. Skin Fibroblasts as the Object for Clinical Diagnosis of Parkinson’s Disease in Persons of Different Ages. Bull. Exp. Biol. Med. 2019, 167, 177–181. [Google Scholar] [CrossRef]
- Khan, T.K.; Nelson, T.J.; Verma, V.A.; Wender, P.A.; Alkon, D.L. A cellular model of Alzheimer’s disease therapeutic efficacy: PKC activation reverses Abeta-induced biomarker abnormality on cultured fibroblasts. Neurobiol. Dis. 2009, 34, 332–339. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic Acid Improves Mitochondrial Function and Redistributes Drp1 in Fibroblasts from Patients with Either Sporadic or Familial Alzheimer’s Disease. J. Mol. Biol. 2018, 430, 3942–3953. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Serrano, G.; Shill, H.A.; Walker, D.G.; Lue, L.; Roher, A.E.; Dugger, B.N.; Maarouf, C.; et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015, 35, 354–389. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Sue, L.I.; Walker, D.G.; Roher, A.E.; Lue, L.; Vedders, L.; Connor, D.J.; Sabbagh, M.N.; Rogers, J. The Sun Health Research Institute Brain Donation Program: Description and experience, 1987-2007. Cell Tissue Bank 2008, 9, 229–245. [Google Scholar] [CrossRef]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beach, T.G.; White, C.L., 3rd; Hladik, C.L.; Sabbagh, M.N.; Connor, D.J.; Shill, H.A.; Sue, L.I.; Sasse, J.; Bachalakuri, J.; Henry-Watson, J.; et al. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol. 2009, 117, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3. research0034.0031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Xie, Y.-Z.; Cao, T.-T.; Wang, Z.; Wang, T.; Li, X.; Shen, R.-C.; Xu, H.; Bu, G.; Chen, X.-F. A rapid and cost-effective method for genotyping apolipoprotein E gene polymorphism. Mol. Neurodegener. 2016, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Polak, U.; Hirsch, C.; Ku, S.; Gottesfeld, J.; Dent, S.Y.; Napierala, M. Selecting and isolating colonies of human induced pluripotent stem cells reprogrammed from adult fibroblasts. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [Green Version]
- Bainbridge, P. Wound healing and the role of fibroblasts. J. Wound Care 2013, 22, 407–408, 410–412. [Google Scholar] [CrossRef]
- Des Jardins-Park, H.E.; Foster, D.S.; Longaker, M.T. Fibroblasts and wound healing: An update. Regen Med. 2018, 13, 491–495. [Google Scholar] [CrossRef] [Green Version]
- Biernaskie, J.; Paris, M.; Morozova, O.; Fagan, B.M.; Marra, M.; Pevny, L.; Miller, F.D. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell 2009, 5, 610–623. [Google Scholar] [CrossRef] [Green Version]
- Hendijani, F. Explant culture: An advantageous method for isolation of mesenchymal stem cells from human tissues. Cell Prolif. 2017, 50. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, L.A.; Sams, M.R.; Deep-Soboslay, A.; Ren-Patterson, R.; Jaffe, A.E.; Chenoweth, J.G.; Jaishankar, A.; Kleinman, J.E.; Hyde, T.M. Use of postmortem human dura mater and scalp for deriving human fibroblast cultures. PLoS ONE 2012, 7, e45282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meske, V.; Albert, F.; Wehser, R.; Ohm, T.G. Culture of autopsy-derived fibroblasts as a tool to study systemic alterations in human neurodegenerative disorders such as Alzheimer’s disease—Methodological investigations. J. Neural. Transm. 1999, 106, 537–548. [Google Scholar] [CrossRef]
- Hjelm, B.E.; Rosenberg, J.B.; Szelinger, S.; Sue, L.I.; Beach, T.G.; Huentelman, M.J.; Craig, D.W. Induction of pluripotent stem cells from autopsy donor-derived somatic cells. Neurosci. Lett. 2011, 502, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Saetern, O.C.; Nguyen, A.; Rodriguez, L.; Schüle, B. Derivation of Leptomeninges Explant Cultures from Postmortem Human Brain Donors. J. Vis. Exp. 2017, 55045. [Google Scholar] [CrossRef]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef] [PubMed]
- Foote, A.G.; Wang, Z.; Kendziorski, C.; Thibeault, S.L. Tissue specific human fibroblast differential expression based on RNAsequencing analysis. BMC Genom. 2019, 20, 308. [Google Scholar] [CrossRef]
- Ishigaki, S.; Fujioka, Y.; Okada, Y.; Riku, Y.; Udagawa, T.; Honda, D.; Yokoi, S.; Endo, K.; Ikenaka, K.; Takagi, S.; et al. Altered Tau Isoform Ratio Caused by Loss of FUS and SFPQ Function Leads to FTLD-like Phenotypes. Cell Rep. 2017, 18, 1118–1131. [Google Scholar] [CrossRef]
- Soundararajan, M.; Kannan, S. Fibroblasts and mesenchymal stem cells: Two sides of the same coin? J. Cell Physiol. 2018, 233, 9099–9109. [Google Scholar] [CrossRef] [PubMed]
- Denu, R.A.; Hematti, P. Effects of Oxidative Stress on Mesenchymal Stem Cell Biology. Oxid Med. Cell Longev 2016, 2016, 2989076. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, G.C.; Singh, P.; Schwarzbauer, J.E. Endogenous production of fibronectin is required for self-renewal of cultured mouse embryonic stem cells. Exp. Cell Res. 2012, 318, 1820–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenselink, E.A. Role of fibronectin in normal wound healing. Int. Wound J. 2015, 12, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Pallari, H.M.; Nevo, J.; Eriksson, J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- Sliogeryte, K.; Gavara, N. Vimentin Plays a Crucial Role in Fibroblast Ageing by Regulating Biophysical Properties and Cell Migration. Cells 2019, 8, 1164. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, N.A.; Tao, R.; Chenoweth, J.G.; Brandtjen, A.; Mighdoll, M.I.; Genova, J.D.; McKay, R.D.; Jia, Y.; Weinberger, D.R.; Kleinman, J.E.; et al. Strong Components of Epigenetic Memory in Cultured Human Fibroblasts Related to Site of Origin and Donor Age. PLoS Genet. 2016, 12, e1005819. [Google Scholar] [CrossRef]
- Fries, K.M.; Blieden, T.; Looney, R.J.; Sempowski, G.D.; Silvera, M.R.; Willis, R.A.; Phipps, R.P. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin. Immunol. Immunopathol. 1994, 72, 283–292. [Google Scholar] [CrossRef]
- Jiang, D.; Rinkevich, Y. Defining Skin Fibroblastic Cell Types Beyond CD90. Front. Cell Dev. Biol. 2018, 6, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korosec, A.; Frech, S.; Gesslbauer, B.; Vierhapper, M.; Radtke, C.; Petzelbauer, P.; Lichtenberger, B.M. Lineage Identity and Location within the Dermis Determine the Function of Papillary and Reticular Fibroblasts in Human Skin. J. Investig. Derm. 2019, 139, 342–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.H.; Zhu, Q.; Li, H.H.; Ra, H.J.; Majumdar, S.; Gulick, D.L.; Jerome, J.A.; Madsen, D.H.; Christofidou-Solomidou, M.; Speicher, D.W.; et al. Fibroblast Activation Protein (FAP) Accelerates Collagen Degradation and Clearance from Lungs in Mice. J. Biol. Chem. 2016, 291, 8070–8089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juillerat-Jeanneret, L.; Tafelmeyer, P.; Golshayan, D. Fibroblast activation protein-α in fibrogenic disorders and cancer: More than a prolyl-specific peptidase? Expert. Opin. Targets 2017, 21, 977–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.E.; Hamson, E.J.; Koczorowska, M.M.; Tholen, S.; Chowdhury, S.; Bailey, C.G.; Lay, A.J.; Twigg, S.M.; Lee, Q.; Roediger, B.; et al. Identification of Novel Natural Substrates of Fibroblast Activation Protein-alpha by Differential Degradomics and Proteomics. Mol. Cell Proteom. 2019, 18, 65–85. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.E.; Ramirez, G.; Hagood, J.S. Roles and regulation of Thy-1, a context-dependent modulator of cell phenotype. Biofactors 2009, 35, 258–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagood, J.S. Thy-1 as an Integrator of Diverse Extracellular Signals. Front. Cell Dev. Biol 2019, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.S.; Reyna, S.; Woodruff, G. Probing the secrets of Alzheimer’s disease using human-induced pluripotent stem cell technology. Neurotherapeutics 2015, 12, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Ghaffari, L.T.; Starr, A.; Nelson, A.T.; Sattler, R. Representing Diversity in the Dish: Using Patient-Derived in Vitro Models to Recreate the Heterogeneity of Neurological Disease. Front. Neurosci. 2018, 12, 56. [Google Scholar] [CrossRef] [Green Version]
- Robbins, J.P.; Price, J. Human induced pluripotent stem cells as a research tool in Alzheimer’s disease. Psychol. Med. 2017, 47, 2587–2592. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.; Ferraiuolo, L.; Miranda, C.J.; Likhite, S.; McElroy, S.; Renusch, S.; Ditsworth, D.; Lagier-Tourenne, C.; Smith, R.A.; Ravits, J.; et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Victor, M.B.; Richner, M.; Hermanstyne, T.O.; Ransdell, J.L.; Sobieski, C.; Deng, P.Y.; Klyachko, V.A.; Nerbonne, J.M.; Yoo, A.S. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 2014, 84, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.L.; Zang, T.; Zhang, C.L. Direct Lineage Reprogramming Reveals Disease-Specific Phenotypes of Motor Neurons from Human ALS Patients. Cell Rep. 2016, 14, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.-S.; Chuang, C.-Y.; Yeh, C.-H.; Chiang, W.; Liu, H.-J.; Lin, T.-N.; Kuo, H.-C. Direct Conversion of Human Fibroblasts into Neural Progenitors Using Transcription Factors Enriched in Human ESC-Derived Neural Progenitors. Stem Cell Rep. 2017, 8, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, T.K.; Alkon, D.L. Early diagnostic accuracy and pathophysiologic relevance of an autopsy-confirmed Alzheimer’s disease peripheral biomarker. Neurobiol. Aging 2010, 31, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, C.; Lilli, C.; Baroni, T.; Parnetti, L.; Sorbi, S.; Emiliani, C.; Lumare, E.; Calabresi, P.; Balloni, S.; Bodo, M. Differences in extracellular matrix production and basic fibroblast growth factor response in skin fibroblasts from sporadic and familial Alzheimer’s disease. Mol. Med. 2007, 13, 542–550. [Google Scholar] [CrossRef]
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast bioenergetics to classify amyotrophic lateral sclerosis patients. Mol. Neurodegener. 2017, 12, 76. [Google Scholar] [CrossRef] [Green Version]
- Raman, R.; Allen, S.P.; Goodall, E.F.; Kramer, S.; Ponger, L.-L.; Heath, P.R.; Milo, M.; Hollinger, H.C.; Walsh, T.; Highley, J.R.; et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 2015, 41, 201–226. [Google Scholar] [CrossRef]
- Riancho, J.; Arozamena, S.; López de Munaín, A. Dermic-derived fibroblasts for the study of amyotrophic lateral sclerosis. Neural Regen Res. 2020, 15, 2043–2044. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables, N | Mean ± S.D. and Ratios | Minimum–Maximum |
|---|---|---|
| Age, N = 29 | 83.6 ± 12.2 years | 43–101 years |
| Gender, N = 29 | 14 males:15 females | |
| APOE genotypes, N = 29 | 2/3;3/3;3/4;4/4 = 3:19:6:1 | |
| Years since diagnosis, N = 21 | 9.0 ± 6.6 years | 1–24 years |
| Age at diagnosis, N = 21 | 71.4 ± 15.1 years | 37–91 years |
| Years of dementia, N = 13 | 4.5 ± 4.0 years | 1–14 years |
| Age of dementia onset, N = 12 | 77.4 ± 10.6 years | 58–92 years |
| Final Diagnosis | MMSE (N) | UPDRS (N) | Sum * of Plaque Density Scores (N) | Sum * of Tangle Density Scores (N) | Sum # of LB Density Scores (N) |
|---|---|---|---|---|---|
| NC | 26.8 ± 3.5 | 12.2 ± 13.4 | 7.8 ± 5.3 | 6.8 ± 2.9 | 2.5 ± 6.1 |
| (6) | (6) | (6) | (6) | (6) | |
| AD | 5.5 ± 0.7 | 30.5 ± 31.8 | 14.3 ± 1.1 | 12.0 ± 0.7 | 14.0 ± 1.4 |
| (2) | (2) | (2) | (2) | (2) | |
| AD/DLB | 5.5 ± 7.8 | 57.5 ± 31.8 | 14.5 ± 0.7 | 10.8 ± 6.0 | 35.5 ± 0.7 |
| (2) | (2) | (2) | (2) | (2) | |
| AD/PSP | 11.0 ± 8.5 | 14.0 | 11.3 ± 1.8 | 8.3 ± 0.4 | 0.0 |
| (2) | (1) | (2) | (2) | (2) | |
| AD/PD | 9.0 | 61.0 | 14.5 ± 0.7 | 5.3 ± 0.4 | 37.5 ± 2.1 |
| (1) | (1) | (2) | (2) | (2) | |
| VAD | 22.0 | 12.0 | 12.5 | 15.0 | 0.0 |
| (1) | (1) | (1) | (1) | (1) | |
| PD | 29.0 | 28.0 | 2.5 | 5.5 | 29.0 |
| (1) | (1) | (1) | (1) | (1) | |
| PDD | NA | NA | 0.0 (1) | 5.0 (1) | 36.0 (1) |
| ALS | 23.0 (1) | NA | 0.0 (1) | 0.5 (1) | 0.0 (1) |
| MSA | 29.0 (1) | NA | 11.0 (1) | 6.5 (1) | 0.0 (1) |
| DNOS | 29.0 | 34.0 | 0.0 | 3.0 | 0.0 |
| (1) | (1) | (1) | (1) | (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beh, S.T.; Frisch, C.; Brafman, D.A.; Churko, J.; Walker, J.E.; Serrano, G.E.; Sue, L.I.; Reiman, E.M.; Beach, T.G.; Lue, L.-F. Human Autopsy-Derived Scalp Fibroblast Biobanking for Age-Related Neurodegenerative Disease Research. Cells 2020, 9, 2383. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112383
Beh ST, Frisch C, Brafman DA, Churko J, Walker JE, Serrano GE, Sue LI, Reiman EM, Beach TG, Lue L-F. Human Autopsy-Derived Scalp Fibroblast Biobanking for Age-Related Neurodegenerative Disease Research. Cells. 2020; 9(11):2383. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112383
Chicago/Turabian StyleBeh, Suet Theng, Carlye Frisch, David A. Brafman, Jared Churko, Jessica E. Walker, Geidy E. Serrano, Lucia I. Sue, Eric M. Reiman, Thomas G. Beach, and Lih-Fen Lue. 2020. "Human Autopsy-Derived Scalp Fibroblast Biobanking for Age-Related Neurodegenerative Disease Research" Cells 9, no. 11: 2383. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112383