Corneal Infection Models: Tools to Investigate the Role of Biofilms in Bacterial Keratitis

 , , , , and
, , , , and
Abstract
:1. Introduction
Predisposing Risk Factors
2. Biofilms
3. Antibiotic Resistance in Biofilms
3.1. Mechanisms of Biofilm-Specific Antibiotic Resistance
3.1.1. Limited Antimicrobial Penetration
3.1.2. The Presence of Altered Chemical Microenvironments
3.1.3. Persister Cells
4. Immune Evasion in Biofilms
4.1. Role of Extracellular Polymeric Substances (EPS)
4.1.1. Mechanical Protection
4.1.2. Immune Recognition
4.2. Changes in Gene Expression
4.3. Manipulation of Host Immune Cells
5. Modelling Biofilm Infections
5.1. In Vitro Models
Existing In Vitro Infection Models
5.2. Ex Vivo Models
Existing Ex Vivo Infection Models
5.3. In Vivo Models
Existing In Vivo Infection Models
6. Conclusions
Funding
Conflicts of Interest
References
- Keay, L.; Edwards, K.; Naduvilath, T.; Taylor, H.R.; Snibson, G.R.; Forde, K.; Stapleton, F. Microbial keratitis—Predisposing factors and morbidity. Ophthalmology 2006, 113, 109–116. [Google Scholar] [CrossRef]
- Keay, L.; Edwards, K.; Stapleton, F. Signs, Symptoms, and Comorbidities in Contact Lens-Related Microbial Keratitis. Optom. Vis. Sci. 2009, 86, 803–809. [Google Scholar] [CrossRef]
- Pascolini, D.; Mariotti, S.P. Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 2012, 96, 614–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitcher, J.P.; Srinivasan, M.; Upadhyay, M.P. Corneal blindness: A global perspective. Bull. World Health Organ. 2001, 79, 214–221. [Google Scholar] [PubMed]
- Collier, S.A.; Gronostaj, M.P.; MacGurn, A.K.; Cope, J.R.; Awsumb, K.L.; Yoder, J.S.; Beach, M.J. Estimated Burden of Keratitis—United States, 2010. Mmwr-Morb. Mortal. Wkly. Rep. 2014, 63, 1027–1030. [Google Scholar]
- Houang, E.; Lam, D.; Fan, D.; Seal, D. Microbial keratitis in Hong Kong: Relationship to climate, environment and contact-lens disinfection. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 361–367. [Google Scholar] [CrossRef]
- Neumann, M.; Sjostrand, J. Central Microbial Keratitis in a Swedish City Population—A 3-Year Prospective-Study in Gothenburg. Acta Ophthalmol. 1993, 71, 160–164. [Google Scholar] [CrossRef]
- Alexandrakis, G.; Alfonso, E.C.; Miller, D. Shifting trends in bacterial keratitis in South Florida and emerging resistance to fluoroquinolones. Ophthalmology 2000, 107, 1497–1502. [Google Scholar] [CrossRef]
- Fleiszig, S.M.J.; Kroken, A.R.; Nieto, V.; Grosser, M.R.; Wan, S.J.; Metruccio, M.M.E.; Evans, D.J. Contact lens-related corneal infection: Intrinsic resistance and its compromise. Prog. Retin. Eye Res. 2019, 100804. [Google Scholar] [CrossRef]
- Bourcier, T.; Thomas, F.; Borderie, V.; Chaumeil, C.; Laroche, L. Bacterial keratitis: Predisposing factors, clinical and microbiological review of 300 cases. Br. J. Ophthalmol. 2003, 87, 834–838. [Google Scholar] [CrossRef] [Green Version]
- Ng, A.L.K.; To, K.K.W.; Choi, C.C.L.; Yuen, L.H.; Yim, S.M.; Chan, K.S.K.; Lai, J.S.M.; Wong, I.Y.H. Predisposing Factors, Microbial Characteristics, and Clinical Outcome of Microbial Keratitis in a Tertiary Centre in Hong Kong: A 10-Year Experience. J. Ophthalmol. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Ung, L.; Bispo, P.J.M.; Shanbhag, S.S.; Gilmore, M.S.; Chodosh, J. The persistent dilemma of microbial keratitis: Global burden, diagnosis, and antimicrobial resistance. Surv. Ophthalmol. 2019, 64, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Bharathi, M.J.; Ramakrishnan, R.; Meenakshi, R.; Padmavathy, S.; Shivakumar, C.; Srinivasan, M. Microbial keratitis in South India: Influence of risk factors, climate, and geographical variation. Ophthalmic Epidemiol. 2007, 14, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Al-Mujaini, A.; Al-Kharusi, N.; Thakral, A.; Wali, U.K. Bacterial keratitis: Perspective on epidemiology, clinico-pathogenesis, diagnosis and treatment. Sultan Qaboos Univ. Med. J. 2009, 9, 184–195. [Google Scholar] [PubMed]
- Chidambaram, J.D.; Prajna, N.V.; Srikanthi, P.; Lanjewar, S.; Shah, M.; Elakkiya, S.; Lalitha, P.; Burton, M.J. Epidemiology, risk factors, and clinical outcomes in severe microbial keratitis in South India. Ophthalmic Epidemiol. 2018, 25, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagerfors, S.; Ejdervik-Lindblad, B.; Soderquist, B. Infectious keratitis: Isolated microbes and their antibiotic susceptibility pattern during 2004-2014 in Region Orebro County, Sweden. Acta Ophthalmol. 2019, 98, 255–260. [Google Scholar] [CrossRef] [Green Version]
- Erie, J.C.; Nevitt, M.P.; Hodge, D.O.; Ballard, D. Incidence of ulcerative keratitis in a defined population from 1950 through 1988. Arch. Ophthalmol. 1993, 111, 1665–1671. [Google Scholar] [CrossRef]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veerachamy, S.; Yarlagadda, T.; Manivasagam, G.; Yarlagadda, P. Bacterial adherence and biofilm formation on medical implants: A review. Proc. Inst. Mech. Eng. Part H-J. Eng. Med. 2014, 228, 1083–1099. [Google Scholar] [CrossRef]
- Zegans, M.E.; Shanks, R.M.Q.; Toole, G.A. Bacterial biofilms and ocular infections. Ocul. Surf. 2005, 3, 73–80. [Google Scholar] [CrossRef]
- Bispo, P.J.M.; Haas, W.; Gilmore, M.S. Biofilms in Infections of the Eye. Pathogens 2015, 4, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Saraswathi, P.; Beuerman, R.W. Corneal Biofilms: From Planktonic to Microcolony Formation in an Experimental Keratitis Infection with Pseudomonas aeruginosa. Ocul. Surf. 2015, 13, 331–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zegans, M.E.; DiGiandomenico, A.; Ray, K.; Naimie, A.; Keller, A.E.; Stover, C.K.; Lalitha, P.; Srinivasan, M.; Acharya, N.R.; Lietman, T.M. Association of Biofilm Formation, Psl Exopolysaccharide Expression, and Clinical Outcomes in Pseudomonas aeruginosa Keratitis Analysis of Isolates in the Steroids for Corneal Ulcers Trial. Jama Ophthalmol. 2016, 134, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dave, A.; Samarth, A.; Karolia, R.; Sharma, S.; Karunakaran, E.; Partridge, L.; MacNeil, S.; Monk, P.N.; Garg, P.; Roy, S. Characterization of Ocular Clinical Isolates of Pseudomonas aeruginosa from Non-Contact Lens Related Keratitis Patients from South India. Microorganisms 2020, 8, 260. [Google Scholar] [CrossRef] [Green Version]
- O’Toole, G.; Kaplan, H.B.; Kolter, R. Biofilm formation as microbial development. Annu. Rev. Microbiol. 2000, 54, 49–79. [Google Scholar] [CrossRef]
- Hoiby, N.; Ciofu, O.; Johansen, H.K.; Song, Z.J.; Moser, C.; Jensen, P.O.; Molin, S.; Givskov, M.; Tolker-Nielsen, T.; Bjarnsholt, T. The clinical impact of bacterial biofilms. Int. J. Oral Sci. 2011, 3, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Stewart, P.; Costerton, J.W. Antibiotic Resistance of Bacteria in Biofilms. Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Hanke, M.L.; Kielian, T. Deciphering mechanisms of staphylococcal biofilm evasion of host immunity. Front. Cell. Infect. Microbiol. 2012, 2, 62. [Google Scholar] [CrossRef] [Green Version]
- Rybtke, M.; Hultqvist, L.D.; Givskov, M.; Tolker-Nielsen, T. Pseudomonas aeruginosa Biofilm Infections: Community Structure, Antimicrobial Tolerance and Immune Response. J. Mol. Biol. 2015, 427, 3628–3645. [Google Scholar] [CrossRef]
- Buhmann, M.T.; Stiefel, P.; Maniura-Weber, K.; Ren, Q. In Vitro Biofilm Models for Device-Related Infections. Trends Biotechnol. 2016, 34, 945–948. [Google Scholar] [CrossRef] [Green Version]
- Cho, P.; Boost, M.V. Evaluation of prevention and disruption of biofilm in contact lens cases. Ophthalmic Physiol. Opt. 2019, 39, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kackar, S.; Suman, E.; Kotian, M.S. Bacterial and Fungal Biofilm formation on Contact Lenses and their Susceptibility to Lens Care Solutions. Indian J. Med. Microbiol. 2017, 35, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Henriques, M.; Sousa, C.; Lira, M.; Elisabete, M.; Oliveira, R.; Azeredo, J. Adhesion of Pseudomonas aeruginosa and Staphylococcus epidermidis to silicone-hydrogel contact lenses. Optom. Vis. Sci. 2005, 82, 446–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.; Cole, N.; Willcox, M. Factors influencing bacterial adhesion to contact lenses. Mol. Vis. 2012, 18, 14–21. [Google Scholar] [PubMed]
- Hsiao, Y.-T.; Fang, P.-C.; Chen, J.-L.; Hsu, S.-L.; Chao, T.-L.; Yu, H.-J.; Lai, Y.-H.; Huang, Y.-T.; Kuo, M.-T. Molecular Bioburden of the Lens Storage Case for Contact Lens-Related Keratitis. Cornea 2018, 37, 1542–1550. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed Res. Int. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Publishes List Of Bacteria for Which New Antibiotics Are Urgently Needed. Available online: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 31 March 2020).
- Tam, A.L.C.; Cote, E.; Saldanha, M.; Lichtinger, A.; Slomovic, A.R. Bacterial Keratitis in Toronto: A 16-Year Review of the Microorganisms Isolated and the Resistance Patterns Observed. Cornea 2017, 36, 1528–1534. [Google Scholar] [CrossRef]
- Chang, V.S.; Dhaliwal, D.K.; Raju, L.; Kowalski, R.P. Antibiotic Resistance in the Treatment of Staphylococcus aureus Keratitis: A 20-Year Review. Cornea 2015, 34, 698–703. [Google Scholar] [CrossRef] [Green Version]
- Tuft, S.; Burton, M. The Royal College Ophthalmologists, Focus: Microbial Keratitis. Available online: https://www.rcophth.ac.uk/wp-content/uploads/2014/08/Focus-Autumn-2013.pdf (accessed on 31 March 2020).
- Saffari, M.; Karami, S.; Firoozeh, F.; Sehat, M. Evaluation of biofilm-specific antimicrobial resistance genes in Pseudomonas aeruginosa isolates in Farabi Hospital. J. Med. Microbiol. 2017, 66, 905–909. [Google Scholar] [CrossRef]
- Heidari, H.; Hadadi, M.; Ebrahim-Saraie, H.S.; Mirzaei, A.; Taji, A.; Hosseini, S.R.; Motamedifar, M. Characterization of virulence factors, antimicrobial resistance patterns and biofilm formation of Pseudomonas aeruginosa and Staphylococcus spp. strains isolated from corneal infection. J. Fr. D’ophtalmol. 2018, 41, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Antibiotic resistance threats in the United States, 2013; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2013; p. 69.
- Kaye, S.; Tuft, S.; Neal, T.; Tole, D.; Leeming, J.; Figueiredo, F.; Armstrong, M.; McDonnell, P.; Tullo, A.; Parry, C. Bacterial Susceptibility to Topical Antimicrobials and Clinical Outcome in Bacterial Keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. [Google Scholar] [CrossRef]
- Anwar, H.; Vanbiesen, T.; Dasgupta, M.; Lam, K.; Costerton, J.W. Interaction of biofilm bacteria with antibiotics in a novel invitro chemostat system. Antimicrob. Agents Chemother. 1989, 33, 1824–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Olsen, I. Biofilm-specific antibiotic tolerance and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 877–886. [Google Scholar] [CrossRef]
- Stewart, P.S. Theoretical aspects of antibiotic diffusion into microbial biofilms. Antimicrob. Agents Chemother. 1996, 40, 2517–2522. [Google Scholar] [CrossRef] [Green Version]
- Mah, T.F.; Pitts, B.; Pellock, B.; Walker, G.C.; Stewart, P.S.; O’Toole, G.A. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature 2003, 426, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Teitzel, G.M.; Balzer, G.J.; Heydorn, A.; Molin, S.; Givskov, M.; Parsek, M.R. Alginate overproduction affects Pseudomonas aeruginosa biofilm structure and function. J. Bacteriol. 2001, 183, 5395–5401. [Google Scholar] [CrossRef] [Green Version]
- Albayaty, Y.N.; Thomas, N.; Hasan, S.; Prestidge, C.A. Penetration of topically used antimicrobials through Staphylococcus aureus biofilms: A comparative study using different models. J. Drug Deliv. Sci. Technol. 2018, 48, 429–436. [Google Scholar] [CrossRef]
- Darouiche, R.O.; Dhir, A.; Miller, A.J.; Landon, G.C.; Raad, I.I.; Musher, D.M. Vancomycin penetration into biofilm covering infected prostheses and effect on bacteria. J. Infect. Dis. 1994, 170, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Sepandj, F.; Ceri, H.; Gibb, A.; Read, R.; Olson, M. Minimum inhibitory concentration (MIC) versus minimum biofilm eliminating concentration (MBEC) in evaluation of antibiotic sensitivity of gram-negative bacilli causing peritonitis. Perit. Dial. Int. 2004, 24, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Tack, K.J.; Sabath, L.D. Increased minimum inhibitory concentrations with anaerobiasis for tobramycin, gentamicin, and amikacin, compared to latamoxef, piperacillin, chloramphenicol, and clindamycin. Chemotherapy 1985, 31, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Bigger, J.W. Treatment of staphylococcal infections with penicillin—By intermittent sterilisation. Lancet 1944, 2, 497–500. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry 2005, 70, 267–274. [Google Scholar] [CrossRef]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [Green Version]
- Askarian, F.; Wagner, T.; Johannessen, M.; Nizet, V. Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol. Rev. 2018, 42, 656–671. [Google Scholar] [CrossRef]
- Kimbrell, D.A.; Beutler, B. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2001, 2, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Rigby, K.M.; DeLeo, F.R. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 2012, 34, 237–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares, E.; Badel-Berchoux, S.; Provot, C.; Prevost, G.; Bernardi, T.; Jehl, F. Clinical Impact of Antibiotics for the Treatment of Pseudomonas aeruginosa Biofilm Infections. Front. Microbiol. 2020, 10, 2894. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Montanaro, L. Implant infections: Adhesion, biofilm formation and immune evasion. Nat. Rev. Microbiol. 2018, 16, 397–409. [Google Scholar] [CrossRef]
- Jensen, P.O.; Givskov, M.; Bjarnsholt, T.; Moser, C. The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol. Med. Microbiol. 2010, 59, 292–305. [Google Scholar] [CrossRef] [Green Version]
- Campoccia, D.; Mirzaei, R.; Montanaro, L.; Arciola, C.R. Hijacking of immune defences by biofilms: A multifront strategy. Biofouling 2019, 35, 1055–1074. [Google Scholar] [CrossRef]
- de Vor, L.; Rooijakkers, S.H.M.; van Strijp, J.A.G. Staphylococci evade the innate immune response by disarming neutrophils and forming biofilms. FEBS Lett. 2020, 594, 2556–2569. [Google Scholar] [CrossRef] [Green Version]
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus Biofilms Prevent Macrophage Phagocytosis and Attenuate Inflammation in vivo. J. Immunol. 2011, 186, 6585–6596. [Google Scholar] [CrossRef] [Green Version]
- Scherr, T.D.; Hanke, M.L.; Huang, O.W.; James, D.B.A.; Horswill, A.R.; Bayles, K.W.; Fey, P.D.; Torres, V.J.; Kielian, T. Staphylococcus aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. MBio 2015, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Herant, M.; Heinrich, V.; Dembo, M. Mechanics of neutrophil phagocytosis: Experiments and quantitative models. J. Cell Sci. 2006, 119, 1903–1913. [Google Scholar] [CrossRef] [Green Version]
- Kovach, K.; Davis-Fields, M.; Irie, Y.; Jain, K.; Doorwar, S.; Vuong, K.; Dhamani, N.; Mohanty, K.; Touhami, A.; Gordon, V.D. Evolutionary adaptations of biofilms infecting cystic fibrosis lungs promote mechanical toughness by adjusting polysaccharide production. Npj Biofilms Microbiomes 2017, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Zapotoczna, M.; McCarthy, H.; Rudkin, J.K.; O’Gara, J.P.; O’Neill, E. An Essential Role for Coagulase in Staphylococcus aureus Biofilm Development Reveals New Therapeutic Possibilities for Device-Related Infections. J. Infect. Dis. 2015, 212, 1883–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristian, S.A.; Birkenstock, T.A.; Sauder, U.; Mack, D.; Gotz, F.; Landmann, R. Biofilm formation induces C3a release and protects Staphylococcus epidermidis from IgG and complement deposition and from neutrophil-dependent killing. J. Infect. Dis. 2008, 197, 1028–1035. [Google Scholar] [CrossRef] [Green Version]
- Cerca, N.; Jefferson, K.K.; Oliveira, R.; Pier, G.B.; Azeredo, J. Comparative antibody-mediated phagocytosis of Staphylococcus epidermidis cells grown in a biofilm or in the planktonic state. Infect. Immun. 2006, 74, 4849–4855. [Google Scholar] [CrossRef] [Green Version]
- Leid, J.G.; Willson, C.J.; Shirtliff, M.E.; Hassett, D.J.; Parsek, M.R.; Jeffers, A.K. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J. Immunol. 2005, 175, 7512–7518. [Google Scholar] [CrossRef] [Green Version]
- Pier, G.B.; Coleman, F.; Grout, M.; Franklin, M.; Ohman, D.E. Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 2001, 69, 1895–1901. [Google Scholar] [CrossRef] [Green Version]
- Mishra, M.; Byrd, M.S.; Sergeant, S.; Azad, A.K.; Parsek, M.R.; McPhail, L.; Schlesinger, L.S.; Wozniak, D.J. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell. Microbiol. 2012, 14, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Novick, R.P. Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol. Microbiol. 2003, 48, 1429–1449. [Google Scholar] [CrossRef]
- He, L.; Le, K.Y.; Khan, B.A.; Nguyen, T.H.; Hunt, R.L.; Bae, J.S.; Kabat, J.; Zheng, Y.; Cheung, G.Y.C.; Li, M.; et al. Resistance to leukocytes ties benefits of quorum sensing dysfunctionality to biofilm infection. Nat. Microbiol. 2019, 4, 1114–1119. [Google Scholar] [CrossRef]
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.L.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.C.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, C.; Saenz, H.L.; Gotz, F.; Otto, M. Impact of the agr quorum-sensing system on adherence to polystyrene in Staphylococcus aureus. J. Infect. Dis. 2000, 182, 1688–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, J.; Rudkin, J.; Recker, M.; Pozzi, C.; O’Gara, J.P.; Massey, R.C. Offsetting virulence and antibiotic resistance costs by MRSA. ISME J. 2010, 4, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Painter, K.L.; Krishna, A.; Wigneshweraraj, S.; Edwards, A.M. What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia? Trends Microbiol. 2014, 22, 676–685. [Google Scholar] [CrossRef]
- Valentini, M.; Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) Signaling: Lessons from Pseudomonas aeruginosa and Other Bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Zhao, K.; Baker, A.E.; Kuchma, S.L.; Coggan, K.A.; Wolfgang, M.C.; Wong, G.C.L.; O’Toole, G.A. A Hierarchical Cascade of Second Messengers Regulates Pseudomonas aeruginosa Surface Behaviors. MBio 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, S.A.; Ahn, D.; Prince, A. Pseudomonas aeruginosa and Klebsiella pneumoniae Adaptation to Innate Immune Clearance Mechanisms in the Lung. J. Innate Immun. 2018, 10, 442–454. [Google Scholar] [CrossRef]
- Cohen, T.S.; Prince, A.S. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J. Clin. Investig. 2013, 123, 1630–1637. [Google Scholar] [CrossRef] [Green Version]
- Huus, K.E.; Joseph, J.; Zhang, L.; Wong, A.; Aaron, S.D.; Mah, T.F.; Sad, S. Clinical Isolates of Pseudomonas aeruginosa from Chronically Infected Cystic Fibrosis Patients Fail To Activate the Inflammasome during Both Stable Infection and Pulmonary Exacerbation. J. Immunol. 2016, 196, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.T.; Kharazmi, A.; Hoiby, N.; Costerton, J.W. Some bacterial parameters influencing the neutrophil oxidative burst response to pseudomonas-aeruginosa biofilms. Apmis 1992, 100, 727–733. [Google Scholar] [CrossRef]
- Guenther, F.; Stroh, P.; Wagner, C.; Obst, U.; Hansch, G.M. Phagocytosis of staphylococci biofilms by polymorphonuclear neutrophils: S. aureus and S. epidermidis differ with regard to their susceptibility towards the host defense. Int. J. Artif. Organs 2009, 32, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Tateda, K.; Ishii, Y.; Horikawa, M.; Matsumoto, T.; Miyairi, S.; Pechere, J.C.; Standiford, T.J.; Ishiguro, M.; Yamaguchi, K. The Pseudomonas aeruginosa autoinducer N-3-oxododecanoyl homoserine lactone accelerates apoptosis in macrophages and neutrophils. Infect. Immun. 2003, 71, 5785–5793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.O.; Bjarnsholt, T.; Phipps, R.; Rasmussen, T.B.; Calum, H.; Christoffersen, L.; Moser, C.; Williams, P.; Pressler, T.; Givskov, M.; et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 2007, 153, 1329–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, K.D.; Foster, S.J.; Renshaw, S.A. Staphylococcus aureus: Setting its sights on the human innate immune system. Microbiology 2019, 165, 367–385. [Google Scholar] [CrossRef]
- Parks, Q.M.; Young, R.L.; Poch, K.R.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: Human F-actin and DNA as targets for therapy. J. Med. Microbiol. 2009, 58, 492–502. [Google Scholar] [CrossRef]
- Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front. Microbiol. 2013, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Robertson, D.M.; Parks, Q.M.; Young, R.L.; Kret, J.; Poch, K.R.; Malcolm, K.C.; Nichols, D.P.; Nichols, M.; Zhu, M.F.; Cavanagh, H.D.; et al. Disruption of Contact Lens-Associated Pseudomonas aeruginosa Biofilms Formed in the Presence of Neutrophils. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2844–2850. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Scott, B.N.V.; Peiseler, M.; Willson, M.E.; Zeng, Z.T.; Warrener, P.; Keller, A.E.; Surewaard, B.G.J.; Dozier, E.A.; Korhonen, J.T.; et al. Neutrophil Extracellular Traps Confine Pseudomonas aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host Microbe 2019, 25, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Berends, E.T.M.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Kockritz-Blickwede, M. Nuclease Expression by Staphylococcus aureus Facilitates Escape from Neutrophil Extracellular Traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [Green Version]
- Winstel, V.; Schneewind, O.; Missiakas, D. Staphylococcus aureus Exploits the Host Apoptotic Pathway To Persist during Infection. MBio 2019, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus Degrades Neutrophil Extracellular Traps to Promote Immune Cell Death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gries, C.M.; Bruger, E.L.; Moormeier, D.E.; Scherr, T.D.; Waters, C.M.; Kielian, T. Cyclic di-AMP Released from Staphylococcus aureus Biofilm Induces a Macrophage Type I Interferon Response. Infect. Immun. 2016, 84, 3564–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, J.F.; Hahn, M.M.; Gunn, J.S. Chronic biofilm-based infections: Skewing of the immune response. Pathog. Dis. 2018, 76, 7. [Google Scholar] [CrossRef] [PubMed]
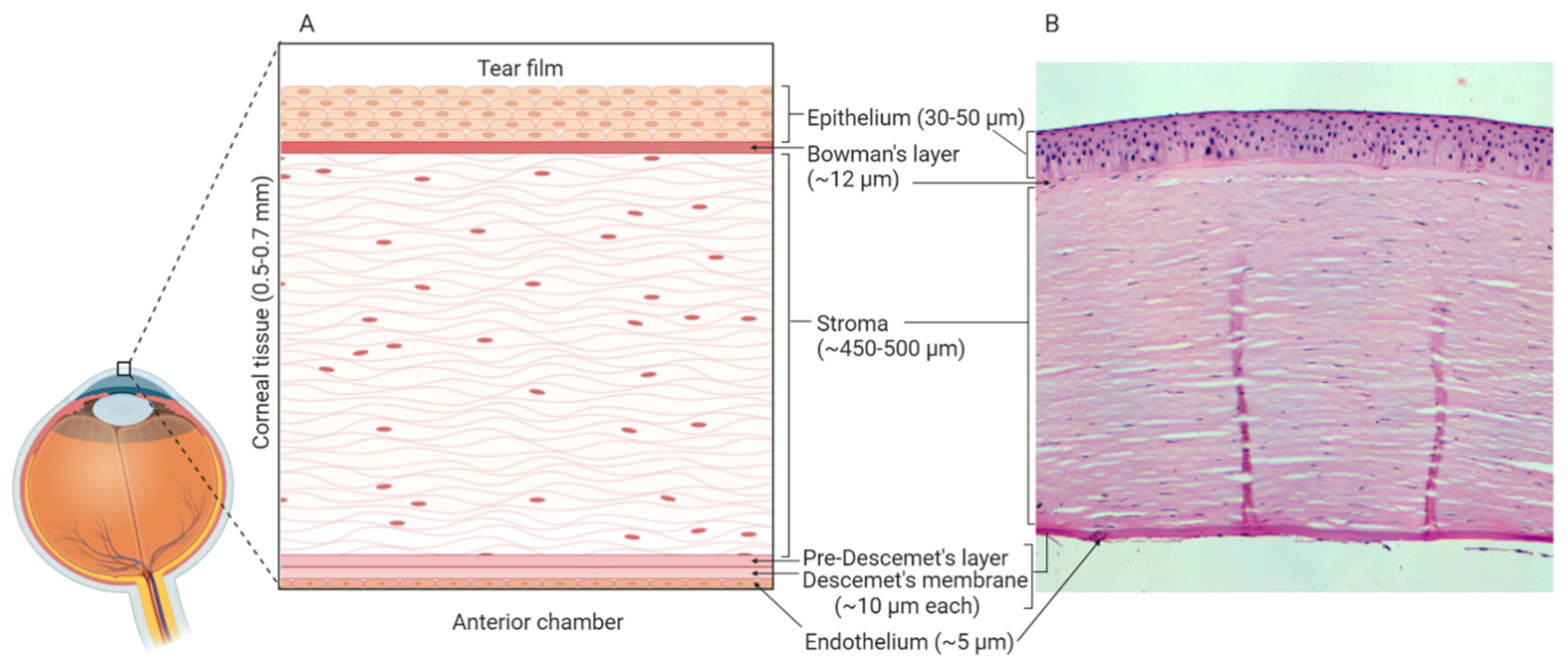
- Krachmer, J.H.; Mannis, M.J.; Holland, E.J. Cornea and Sclera: Anatomy and Physiology. In Cornea, 3rd ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2010; pp. 3–15. [Google Scholar]
- Dua, H.S.; Faraj, L.A.; Said, D.G.; Gray, T.; Lowe, J. Human Corneal Anatomy Redefined A Novel Pre-Descemet’s Layer (Dua’s Layer). Ophthalmology 2013, 120, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Sosnova-Netukova, M.; Kuchynka, P.; Forrester, J.V. The suprabasal layer of corneal epithelial cells represents the major barrier site to the passive movement of small molecules and trafficking leukocytes. Br. J. Ophthalmol. 2007, 91, 372–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranta, V.P.; Laavola, M.; Toropainen, E.; Vellonen, K.S.; Talvitie, A.; Urtti, A. Ocular pharmacokinetic modeling using corneal absorption and desorption rates from in vitro permeation experiments with cultured corneal epithelial cells. Pharm. Res. 2003, 20, 1409–1416. [Google Scholar] [CrossRef]
- Toropainen, E. Corneal Epithelial Cell Culture Model for Pharmaceutical Studies. Ph.D. Dissertation, University of Kuopio, Kuopio, Finland, 2007. [Google Scholar]
- Kaluzhny, Y.; Kinuthia, M.W.; Truong, T.; Lapointe, A.M.; Hayden, P.; Klausner, M. New Human Organotypic Corneal Tissue Model for Ophthalmic Drug Delivery Studies. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2880–2898. [Google Scholar] [CrossRef] [Green Version]
- Zorn-Kruppa, M.; Tykhonova, S.; Belge, G.; Bednarz, J.; Diehl, H.A.; Engelke, M. A human corneal equivalent constructed from SV40-immortalised corneal cell lines. Atla-Altern. Lab. Anim. 2005, 33, 37–45. [Google Scholar] [CrossRef]
- Builles, N.; Bechetoille, N.; Justin, V.; Andre, V.; Barbaro, V.; Di Iorio, E.; Auxenfans, C.; Hulmes, D.J.S.; Damour, O. Development of a hemicornea from human primary cell cultures for pharmacotoxicology testing. Cell Biol. Toxicol. 2007, 23, 279–292. [Google Scholar] [CrossRef]
- Kahn, C.R.; Young, E.; Lee, I.H.; Rhim, J.S. Human corneal epithelial primary cultures and cell-lines with extended life-span-in-vitro model for ocular studies. Investig. Ophthalmol. Vis. Sci. 1993, 34, 3429–3441. [Google Scholar]
- Ouellette, M.M.; McDaniel, L.D.; Wright, W.E.; Shay, J.W.; Schultz, R.A. The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum. Mol. Genet. 2000, 9, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Greco, D.; Vellonen, K.S.; Turner, H.C.; Hakli, M.; Tervo, T.; Auvinen, P.; Wolosin, J.M.; Urtti, A. Gene expression analysis in SV-40 immortalized human corneal epithelial cells cultured with an air-liquid interface. Mol. Vis. 2010, 16, 2109–2120. [Google Scholar] [PubMed]
- Postnikoff, C.K.; Pintwala, R.; Williams, S.; Wright, A.M.; Hileeto, D.; Gorbet, M.B. Development of a Curved, Stratified, In Vitro Model to Assess Ocular Biocompatibility. PLoS ONE 2014, 9, e96448. [Google Scholar] [CrossRef] [PubMed]
- Toropainen, E.; Ranta, V.P.; Talvitie, A.; Suhonen, P.; Urtti, A. Culture model of human corneal epithelium for prediction of ocular drug absorption. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2942–2948. [Google Scholar]
- Toropainen, E.; Ranta, V.P.; Vellonen, K.S.; Palmgren, J.; Talvitie, A.; Laavola, M.; Suhonen, P.; Hamalainen, K.M.; Auriola, S.; Urtti, A. Paracellular and passive transcellular permeability in immortalized human corneal epithelial cell culture model. Eur. J. Pharm. Sci. 2003, 20, 99–106. [Google Scholar] [CrossRef]
- Reichl, S.; Kolln, C.; Hahne, M.; Verstraelen, J. In vitro cell culture models to study the corneal drug absorption. Expert Opin. Drug Metab. Toxicol. 2011, 7, 559–578. [Google Scholar] [CrossRef]
- Dey, S. Corneal cell culture models: A tool to study corneal drug absorption. Expert Opin. Drug Metab. Toxicol. 2011, 7, 529–532. [Google Scholar] [CrossRef]
- Jett, B.D.; Gilmore, M.S. Internalization of Staphylococcus aureus by human corneal epithelial cells: Role of bacterial fibronectin-binding protein and host cell factors. Infect. Immun. 2002, 70, 4697–4700. [Google Scholar] [CrossRef] [Green Version]
- Garcia, B.; Merayo-Lloves, J.; Rodriguez, D.; Alcalde, I.; Garcia-Suarez, O.; Alfonso, J.F.; Baamonde, B.; Fernandez-Vega, A.; Vazquez, F.; Quiros, L.M. Different Use of Cell Surface Glycosaminoglycans As Adherence Receptors to Corneal Cells by Gram Positive and Gram Negative Pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 173. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Guha, S.; Garg, P.; Roy, S. Differential expression of antimicrobial peptides in corneal infection and regulation of antimicrobial peptides and reactive oxygen species by type III secretion system of &ITPseudomonas aeruginosa & IT. Pathog. Dis. 2018, 76, fty001. [Google Scholar] [CrossRef] [Green Version]
- Gipson, I.K.; Spurr-Michaud, S.; Tisdale, A.; Menon, B.B. Comparison of the Transmembrane Mucins MUC1 and MUC16 in Epithelial Barrier Function. PLoS ONE 2014, 9, e100393. [Google Scholar] [CrossRef] [PubMed]
- Fleiszig, S.M.J.; Kwong, M.S.F.; Evans, D.J. Modification of Pseudomonas aeruginosa interactions with corneal epithelial cells by human tear fluid. Infect. Immun. 2003, 71, 3866–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, M.S.F.; Evans, D.J.; Ni, M.; Cowell, B.A.; Fleiszig, S.M.J. Human tear fluid protects against Pseudomonas aeruginosa keratitis in a murine experimental model. Infect. Immun. 2007, 75, 2325–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponce-Angulo, D.G.; Bautista-Hernandez, L.A.; Calvillo-Medina, R.P.; Castro-Tecorral, F.I.; Aparicio-Ozores, G.; Lopez-Villegas, E.O.; Ribas-Aparicio, R.M.; Bautista-de Lucio, V.M. Microscopic characterization of biofilm in mixed keratitis in a novel murine model. Microb. Pathog. 2020, 140, 103953. [Google Scholar] [CrossRef]
- Doroshenko, N.; Rimmer, S.; Hoskins, R.; Garg, P.; Swift, T.; Spencer, H.L.M.; Lord, R.M.; Katsikogianni, M.; Pownall, D.; MacNeil, S.; et al. Antibiotic functionalised polymers reduce bacterial biofilm and bioburden in a simulated infection of the cornea. Biomater. Sci. 2018, 6, 2101–2109. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.E.L.; Kragh, K.N.; Bjarnsholt, T.; Diggle, S.P. The Limitations of In Vitro Experimentation in Understanding Biofilms and Chronic Infection. J. Mol. Biol. 2015, 427, 3646–3661. [Google Scholar] [CrossRef]
- Palmer, K.L.; Aye, L.A.; Whiteley, M. Nutritional cues control Pseudomonas aeruginosa multicellular Behavior in cystic fibrosis sputum. J. Bacteriol. 2007, 189, 8079–8087. [Google Scholar] [CrossRef] [Green Version]
- Bjarnsholt, T.; Alhede, M.; Eickhardt-Sorensen, S.R.; Moser, C.; Kuhl, M.; Jensen, P.O.; Hoiby, N. The in vivo biofilm. Trends Microbiol. 2013, 21, 466–474. [Google Scholar] [CrossRef]
- Kolpen, M.; Bjarnsholt, T.; Moser, C.; Hansen, C.R.; Rickelt, L.F.; Kuhl, M.; Hempel, C.; Pressler, T.; Hoiby, N.; Jensen, P.O. Nitric oxide production by polymorphonuclear leucocytes in infected cystic fibrosis sputum consumes oxygen. Clin. Exp. Immunol. 2014, 177, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Metruccio, M.M.E.; Tam, C.; Evans, D.J.; Xie, A.L.; Stern, M.E.; Fleiszig, S.M.J. Contributions of MyD88-dependent receptors and CD11c-positive cells to corneal epithelial barrier function against Pseudomonas aeruginosa. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Metruccio, M.M.E.; Wan, S.J.; Horneman, H.; Kroken, A.R.; Sullivan, A.B.; Truong, T.N.; Mun, J.J.; Tam, C.K.P.; Frith, R.; Welsh, L.; et al. A novel murine model for contact lens wear reveals clandestine IL-1R dependent corneal parainflammation and susceptibility to microbial keratitis upon inoculation with Pseudomonas aeruginosa. Ocul. Surf. 2019, 17, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.B.; Tam, K.P.C.; Metruccio, M.M.E.; Evans, D.J.; Fleiszig, S.M.J. The Importance of the Pseudomonas aeruginosa Type III Secretion System in Epithelium Traversal Depends upon Conditions of Host Susceptibility. Infect. Immun. 2015, 83, 1629–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Liu, C.L.; Liu, S.H.; Cong, H.J.; Chen, Y.H.; Gu, L.C.; Ma, L.Y.Z. Membrane association of SadC enhances its diguanylate cyclase activity to control exopolysaccharides synthesis and biofilm formation in Pseudomonas aeruginosa. Environ. Microbiol. 2016, 18, 3440–3452. [Google Scholar] [CrossRef]
- Zhu, H.; Kochevar, I.E.; Behlau, I.; Zhao, J.; Wang, F.H.; Wang, Y.C.; Sun, X.D.; Hamblin, M.R.; Dai, T.H. Antimicrobial Blue Light Therapy for Infectious Keratitis: Ex Vivo and In Vivo Studies. Investig. Ophthalmol. Vis. Sci. 2017, 58, 586–593. [Google Scholar] [CrossRef] [Green Version]
- Hume, E.B.H.; Dajcs, J.J.; Moreau, J.M.; Sloop, G.D.; Willcox, M.D.P.; O’Callaghan, R.J. Staphylococcus corneal virulence in a new topical model of infection. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2904–2908. [Google Scholar]
- Pinnock, A.; Shivshetty, N.; Roy, S.; Rimmer, S.; Douglas, I.; MacNeil, S.; Garg, P. Ex vivo rabbit and human corneas as models for bacterial and fungal keratitis. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Lawinbrussel, C.A.; Refojo, M.F.; Leong, F.L.; Hanninen, L.; Kenyon, K.R. Effect of pseudomonas-aeruginosa concentration in experimental contact lens-related microbial keratitis. Cornea 1993, 12, 10–18. [Google Scholar] [CrossRef]
- Ren, H.; Petroll, W.; Jester, J.; Cavanagh, H.; Mathers, W.; Bonnano, J.; Kennedy, R. Adherence of Pseudomonas aeruginosa to shed rabbit corneal epithelial cells after overnight wear of contact lenses. Contact Lens Assoc. Ophthalmol. J. 1997, 23, 63–68. [Google Scholar]
- Robertson, D.M.; Rogers, N.A.; Petroll, W.M.; Zhu, M.F. Second harmonic generation imaging of corneal stroma after infection by Pseudomonas aeruginosa. Sci. Rep. 2017, 7, 46116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhu, S.N.; Jha, K.K.; Karthyayani, A.P.; Gajjar, D.U. Ex vivo Caprine Model to Study Virulence Factors in Keratitis. J. Ophthalmic Vis. Res. 2018, 13, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.S.; Hu, F.R.; Chen, C.T. Photodynamic antimicrobial chemotherapy for methicillin-resistant Staphylococcus aureus -in vitro, biofilm, and ex vivo bovine keratitis model. Investig. Ophthalmol. Vis. Sci. 2013, 54. [Google Scholar]
- Vermeltfoort, P.B.J.; van Kooten, T.G.; Bruinsma, G.M.; Hooymans, A.M.M.; van der Mei, H.C.; Busscher, H.J. Bacterial transmission from contact lenses to porcine corneas: An ex vivo study. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2042–2046. [Google Scholar] [CrossRef] [PubMed]
- Brothers, K.M.; Stella, N.A.; Hunt, K.M.; Romanowski, E.G.; Liu, X.Y.; Klarlund, J.K.; Shanks, R.M.Q. Putting on the brakes: Bacterial impediment of wound healing. Sci. Rep. 2015, 5, 14003. [Google Scholar] [CrossRef] [PubMed]
- Okurowska, K.; Roy, S.; Thokala, P.; Partridge, L.; Garg, P.; MacNeil, S.; Monk, P.N.; Karunakaran, E. Establishing a Porcine Ex Vivo Cornea Model for Studying Drug Treatments against Bacterial Keratitis. J. Vis. Exp. 2020, 159. [Google Scholar] [CrossRef]
- Agarwal, P.; Rupenthal, I.D. In vitro and ex vivo corneal penetration and absorption models. Drug Deliv. Transl. Res. 2016, 6, 634–647. [Google Scholar] [CrossRef]
- Hatami-Marbini, H.; Etebu, E.; Rahimi, A. Swelling Pressure and Hydration Behavior of Porcine Corneal Stroma. Curr. Eye Res. 2013, 38, 1124–1132. [Google Scholar] [CrossRef]
- Ehlers, N.; Heegaard, S.; Hjortdal, J.; Ivarsen, A.; Nielsen, K.; Prause, J.U. Morphological evaluation of normal human corneal epithelium. Acta Ophthalmol. 2010, 88, 858–861. [Google Scholar] [CrossRef]
- Abhari, S.; Eisenback, M.; Kaplan, H.J.; Walters, E.; Prather, R.S.; Scott, P.A. Anatomic Studies of the Miniature Swine Cornea. Anat. Rec. -Adv. Integr. Anat. Evol. Biol. 2018, 301, 1955–1967. [Google Scholar] [CrossRef] [Green Version]
- Marquart, M.E. Animal Models of Bacterial Keratitis. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Merindano, M.D.; Costa, J.; Canals, M.; Potau, J.M.; Ruano, D. A comparative study of Bowman’s layer in some mammals: Relationships with other constituent corneal structures. Eur. J. Anat. 2002, 6, 133–139. [Google Scholar]
- Wilson, S.E. Bowman’s layer in the cornea– structure and function and regeneration. Exp. Eye Res. 2020, 195. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, I.; Kwan, L.; Yu, C.; Evans, D.J.; Fleiszig, S.M.J. Role of the Corneal Epithelial Basement Membrane in Ocular Defense against Pseudomonas aeruginosa. Infect. Immun. 2009, 77, 3264–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, L.; Brocas, A.; Singh, K.; Kieffer, J.C.; Brunette, I.; Ozaki, T. Determination of porcine corneal layers with high spatial resolution by simultaneous second and third harmonic generation microscopy. Opt. Express 2008, 16, 16284–16293. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Moral, M.; Garcia-Posadas, L.; Lopez-Garcia, A.; Diebold, Y. Histological and immunohistochemical characterization of the porcine ocular surface. PLoS ONE 2020, 15, e0227732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, A.; Breunig, H.G.; Uchugonova, A.; Morgado, A.M.; Konig, K. Two-photon spectral fluorescence lifetime and second-harmonic generation imaging of the porcine cornea with a 12-femtosecond laser microscope. J. Biomed. Opt. 2016, 21, 036002. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, J.L.; Ventosa, J.A.; Piedra, S. The three-dimensional microanatomy of the rabbit and human cornea. A chemical and mechanical microdissection-SEM approach. J. Anat. 2001, 199, 567–576. [Google Scholar] [CrossRef]
- Lai, T.; Tang, S. Cornea characterization using a combined multiphoton microscopy and optical coherence tomography system. Biomed. Opt. Express 2014, 5, 1494–1511. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Osawa, T.; Tohyama, K. Comparative observations on corneas, with special reference to Bowman’s layer and Descemet’s membrane in mammals and amphibians. J. Morphol. 2002, 254, 247–258. [Google Scholar] [CrossRef]
- Ramphal, R.; McNiece, M.T.; Polack, F.M. Adherence Of Pseudomonas-Aeruginosa to the Injured Cornea—A Step In The Pathogenesis Of Corneal Infections. Ann. Ophthalmol. 1981, 13, 421–425. [Google Scholar] [PubMed]
- Vallas, V.; WienerKronish, J.P.; Mostov, K.E.; Fleiszig, S.M.J. Cytotoxic strains of Pseudomonas aeruginosa can damage the intact corneal surface. Investig. Ophthalmol. Vis. Sci. 1996, 37, 4026. [Google Scholar]
- Klotz, S.A.; Au, Y.K.; Misra, R.P. A Partial-Thickness Epithelial Defect Increases the Adherence of Pseudomonas-Aeruginosa to the Cornea. Investig. Ophthalmol. Vis. Sci. 1989, 30, 1069–1074. [Google Scholar]
- Augustin, D.K.; Heimer, S.R.; Tam, C.; Li, W.Y.; Le Due, J.M.; Evans, D.J.; Fleiszig, S.M.J. Role of Defensins in Corneal Epithelial Barrier Function against Pseudomonas aeruginosa Traversal. Infect. Immun. 2011, 79, 595–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubani-Ukoma, U.; Gibson, D.; Schultz, G.; Silva, B.O.; Chauhan, A. Evaluating the potential of drug eluting contact lenses for treatment of bacterial keratitis using an ex vivo corneal model. Int. J. Pharm. 2019, 565, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.; Mun, J.J.; Evans, D.J.; Fleiszig, S.M.J. The Impact of Inoculation Parameters on the Pathogenesis of Contact Lens-Related Infectious Keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3100–3106. [Google Scholar] [CrossRef]
- Jian, H.J.; Yu, J.T.; Li, Y.J.; Unnikrishnan, B.; Huang, Y.F.; Luo, L.J.; Ma, D.H.K.; Harroun, S.G.; Chang, H.T.; Lin, H.J.; et al. Highly adhesive carbon quantum dots from biogenic amines for prevention of biofilm formation. Chem. Eng. J. 2020, 386, 123913. [Google Scholar] [CrossRef]
- Tang, A.H.; Caballero, A.R.; Marquart, M.E.; Bierdeman, M.A.; O’Callaghan, R.J. Mechanism of Pseudomonas aeruginosa Small Protease (PASP), a Corneal Virulence Factor. Investig. Ophthalmol. Vis. Sci. 2018, 59, 5993–6002. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, M.; Barathi, V.A.; Goh, E.T.L.; Anggara, R.; Fazil, M.; Ng, A.J.Y.; Harini, S.; Aung, T.T.; Fox, S.J.; Liu, S.P.; et al. Antimicrobial Activity and Cell Selectivity of Synthetic and Biosynthetic Cationic Polymers. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ma, X.; Zhao, L.; Li, Y.; Zhou, Q.; Du, X. Extended contact lens wear promotes corneal norepinephrine secretion and Pseudomonas aeruginosa infection in mice. Investig. Ophthalmol Vis. Sci. 2020, 61, 17. [Google Scholar] [CrossRef]
- Kugadas, A.; Geddes-McAlister, J.; Guy, E.; DiGiandomenico, A.; Sykes, D.B.; Mansour, M.K.; Mirchev, R.; Gadjeva, M. Frontline Science: Employing enzymatic treatment options for management of ocular biofilm-based infections. J. Leukoc. Biol. 2019, 105, 1099–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saraswathi, P.; Aung, T.; Salleh, S.; Beuerman, R. An Experimental Model of Biofilm Formation in the Mouse Cornea. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5208. [Google Scholar]
- Metruccio, M.M.E.; Evans, D.J.; Gabriel, M.M.; Kadurugamuwa, J.L.; Fleiszig, S.M.J. Pseudomonas aeruginosa Outer Membrane Vesicles Triggered by Human Mucosal Fluid and Lysozyme Can Prime Host Tissue Surfaces for Bacterial Adhesion. Front. Microbiol. 2016, 7, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksson, J.T.; McDermott, A.M.; Bergmanson, J.P.G. Dimensions and Morphology of the Cornea in Three Strains of Mice. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3648–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in Innate Immune Response between Man and Mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.; LeDue, J.; Mun, J.J.; Herzmark, P.; Robey, E.A.; Evans, D.J.; Fleiszig, S.M.J. 3D Quantitative Imaging of Unprocessed Live Tissue Reveals Epithelial Defense against Bacterial Adhesion and Subsequent Traversal Requires MyD88. PLoS ONE 2011, 6, e24008. [Google Scholar] [CrossRef]
- Mun, J.J.; Tam, C.; Kowbel, D.; Hawgood, S.; Barnett, M.J.; Evans, D.J.; Fleiszig, S.M.J. Clearance of Pseudomonas aeruginosa from a Healthy Ocular Surface Involves Surfactant Protein D and Is Compromised by Bacterial Elastase in a Murine Null-Infection Model. Infect. Immun. 2009, 77, 2392–2398. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.J.; Ma, S.; Evans, D.J.; Fleiszig, S.M.J. Resistance of the murine cornea to bacterial colonization during experimental dry eye. PLoS ONE 2020, 15, e0234013. [Google Scholar] [CrossRef]
- Yeung, J.; Gadjeva, M.; Geddes-McAlister, J. Label-Free Quantitative Proteomics Distinguishes General and Site-Specific Host Responses to Pseudomonas aeruginosa Infection at the Ocular Surface. Proteomics 2020, 20, 1900290. [Google Scholar] [CrossRef]
- Sewell, A.; Dunmire, J.; Wehmann, M.; Rowe, T.; Bouhenni, R. Proteomic analysis of keratitis-associated Pseudomonas aeruginosa. Mol. Vis. 2014, 20, 1182–1191. [Google Scholar]
- Sun, Y.; Karmakar, M.; Roy, S.; Ramadan, R.T.; Williams, S.R.; Howell, S.; Shive, C.L.; Han, Y.P.; Stopford, C.M.; Rietsch, A.; et al. TLR4 and TLR5 on Corneal Macrophages Regulate Pseudomonas aeruginosa Keratitis by Signaling through MyD88-Dependent and -Independent Pathways. J. Immunol. 2010, 185, 4272–4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Okamoto, S.; Oka, N.; Hayashi, N.; Gotoh, N.; Shiraishi, A. Role of pvdE Pyoverdine Synthesis in Pseudomonas aeruginosa Keratitis. Cornea 2018, 37, S99–S105. [Google Scholar] [CrossRef] [PubMed]
- Clemens, L.E.; Jaynes, J.; Lim, E.; Kolar, S.S.; Reins, R.Y.; Baidouri, H.; Hanlon, S.; McDermott, A.M.; Woodburn, K.W. Designed Host Defense Peptides for the Treatment of Bacterial Keratitis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6273–6281. [Google Scholar] [CrossRef] [PubMed]
- Ni, M.J.; Tam, C.; Verma, A.; Ramphal, R.; Hawgood, S.; Evans, D.J.; Fleiszig, S.M.J. Expression of surfactant protein D in human corneal epithelial cells is upregulated by Pseudomonas aeruginosa. FEMS Immunol. Med. Microbiol. 2008, 54, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.Y.; McClellan, S.A.; Barrett, R.P.; Zhang, Y.F.; Foldenauer, M.E.; Hazlett, L.D. The Role of VIP in Cornea. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7560–7566. [Google Scholar] [CrossRef] [PubMed]
- Barbariga, M.; Vallone, F.; Mosca, E.; Bignami, F.; Magagnotti, C.; Fonteyne, P.; Chiappori, F.; Milanesi, L.; Rama, P.; Andolfo, A.; et al. The role of extracellular matrix in mouse and human corneal neovascularization. Sci. Rep. 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Advantages | Disadvantages | |
|---|---|---|
| in vitrocell culture models | ■ Economical. ■ Reduced use of animals. ■ Cell lines can be used continuously. ■ 3D organotypic models can be developed using multiple cell lines. ■ Many host defence mechanisms remain investigable, e.g. expression of mucins, AMPs, pro-inflammatory cytokines and microRNAs, investigation of cell surface receptors and PRR signalling pathways. | ■ Problems with cell lines and genetic drift. ■ Primary cells reach senescence after a few passages. ■ Reduced cell viability and increased susceptibility to infection. ■ Absence of resident and infiltrative immune cells. ■ Absence of conjunctiva. ■ Absence of tear fluid and lacrimal glands. ■ Infection normally occurs under static conditions. ■ Differences in the biofilm microenvironment (e.g. nutritional cues, absence of immune cells) may affect biofilm morphology. |
| ex vivomodels | ■ Whole-tissue model. ■ Complex 3D surface topology of the cornea is preserved. ■ Increased cell viability facilitates longer infection periods. ■ Presence of resident immune cells. | ■ Low availability of human corneas means animal models are commonly used. ■ Lack of standardised infection methods. ■ Dispute regarding corneal anatomy of animal models. ■ Interspecies differences in corneal anatomy, functional characteristics and immune response may affect applicability to human infections. ■ Absence of infiltrative immune cells. ■ Absence of conjunctiva. ■ Absence of tear fluid and lacrimal glands. ■ Infection normally occurs under static conditions. ■ Differences in the biofilm microenvironment (e.g. nutritional cues, absence of immune cells) may affect biofilm morphology. |
| in vivomodels | ■ Complete immune response (resident/infiltrative immune cells, tear film, conjunctiva and lymphatic vessels). ■ Infection occurs under dynamic, shear stress conditions. ■ Biofilm morphology should be highly similar to the true infectious scenario. | ■ Animal models must be used, raising ethical issues. ■ Interspecies differences in corneal anatomy, functional characteristics and immune response may affect applicability to human infections. ■ Expensive. ■ Time-consuming. ■ Infections can be difficult to establish and prior wounding of the cornea is often required. |
| Animal model | Pathogen | Biofilm Characteristics | Ref. |
|---|---|---|---|
| C57BL/6 black mice | Pseudomonas aeruginosa ATCC 9027 | ■ Rapid shift from planktonic to biofilm lifestyle observed for all corneas. ■ Microcolonies present on day 2 post-infection and fibrous extracellular substances visible. ■ Mature biofilm structures present on day 3. Bacteria form as “mushroom shaped bodies” and “tower like structures” and are embedded in a web of extracellular polysaccharides. ■ A thick, dense biofilm layer is observed on days 5-6. Bacteria become static within this structure. ■ Neutrophils migrate into the corneal stroma and production of NETs is observed at early time points. Neutrophils are localised to the biofilm surface once mature biofilm structures develop. | [22] |
| C57BL/6 and Swiss Webster (SW) mice | Pseudomonas aeruginosa PAO1-GFP and 6294-GFP (clinical isolate) | ■ Early (12 h) biofilms are composed of bacterial clusters/microcolonies that are thought to emanate from the infected epithelial cells. ■ Late (24 h) biofilms are composed of bacterial sheets. ■ Biofilm bacteria are surrounded by Psl polysaccharide but there is a low abundance of alginate. ■ Biofilms are resistant to neutrophil infiltration. | [176] |
| BALB/c mice | Staphylococcus aureus and Fusarium falciforme (clinical isolates) | ■ A mixed biofilm is observed after 72 h. ■ S. aureus: Bacteria colonise the corneal epithelium and a part of the stroma. Bacterial clusters observed, including a large cocci aggregate at the site of the corneal lesion. Bacteria secrete exopolysaccharides that form “halos” around the bacteria and then merge with the extracellular matrix of other cocci. Development of a new blood vessel in the stroma is observed and attributed to the host immune response. ■ F. falciforme: Hyphae and conidia observed and hyphae migrates through stroma to reach the endothelium. F. falciforme structures are embedded in a fibrin matrix within the stroma. Presence/growth of fungi causes corneal collagen fibres to become disorganised. | [131] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urwin, L.; Okurowska, K.; Crowther, G.; Roy, S.; Garg, P.; Karunakaran, E.; MacNeil, S.; Partridge, L.J.; Green, L.R.; Monk, P.N. Corneal Infection Models: Tools to Investigate the Role of Biofilms in Bacterial Keratitis. Cells 2020, 9, 2450. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112450
Urwin L, Okurowska K, Crowther G, Roy S, Garg P, Karunakaran E, MacNeil S, Partridge LJ, Green LR, Monk PN. Corneal Infection Models: Tools to Investigate the Role of Biofilms in Bacterial Keratitis. Cells. 2020; 9(11):2450. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112450
Chicago/Turabian StyleUrwin, Lucy, Katarzyna Okurowska, Grace Crowther, Sanhita Roy, Prashant Garg, Esther Karunakaran, Sheila MacNeil, Lynda J. Partridge, Luke R. Green, and Peter N. Monk. 2020. "Corneal Infection Models: Tools to Investigate the Role of Biofilms in Bacterial Keratitis" Cells 9, no. 11: 2450. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9112450