Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs)

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Generation of Nb-CARs
2.3. Stable Transduction of NK-92 Cells with Nb-CARs
2.4. Production of Alexa Fluor 647-Labeled CD38 and ARTC2.2
2.5. Luminescence CARDCC Assays
2.6. Flow Cytometric CARDCC Assays
2.7. CARDCC Assays with Primary Human Bone Marrow Samples
3. Results
3.1. Generation of CD38-Deficient Cell Lines and Lentiviral Transduction of CD38+ and CD38− Cell Lines with Luciferase
3.2. Generation of Nanobody-Based Chimeric Antigen Receptors (Nb-CARs) and Transduction of NK-92 Cells with Nb-CARs
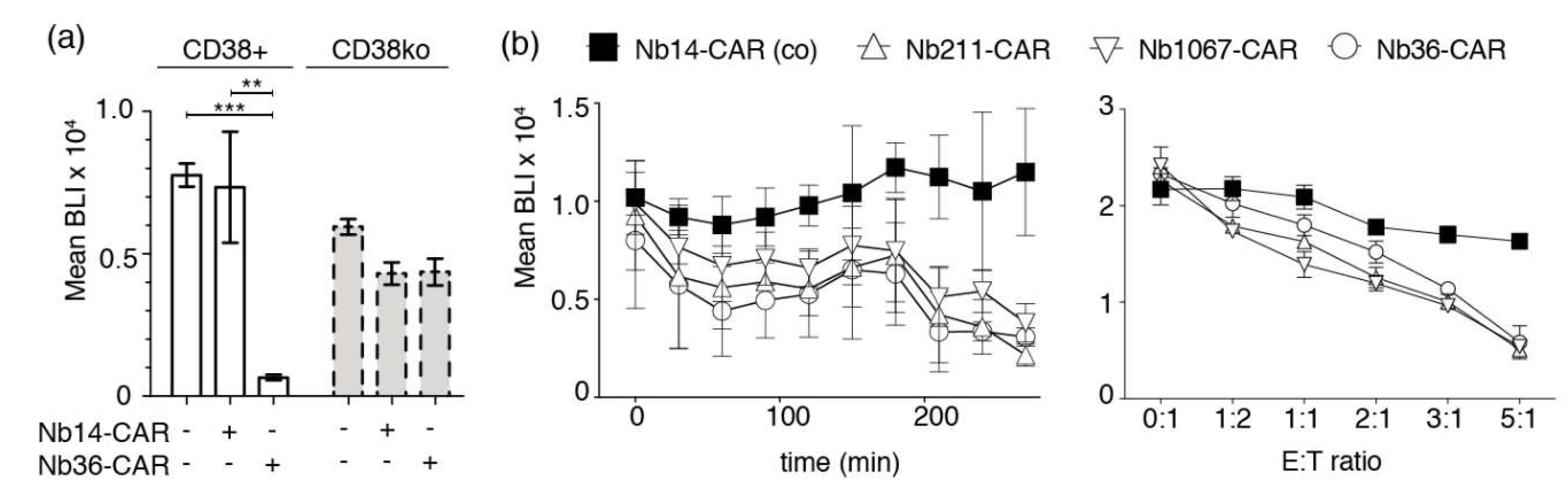
3.3. Luminescence-Based CAR-Dependent Cellular Cytotoxicity Assay (CARDCC)
3.4. A Flow-Cytometric CARDCC
3.5. CD38-Directed Nb-CAR-NKs Specifically Deplete CD38+/CD56+ Myeloma Cells from Primary Human Bone Marrow Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deaglio, S.; Aydin, S.; Vaisitti, T.; Bergui, L.; Malavasi, F. CD38 at the junction between prognostic marker and therapeutic target. Trends Mol. Med. 2008, 14, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in adenosinergic pathways and metabolic re-programming in human multiple myeloma cells: In-tandem insights from basic science to therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A target for immunotherapeutic approaches in multiple myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.W.; Comenzo, R.L. CD38 Monoclonal Antibody Therapies for Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2015, 15, 635–645. [Google Scholar] [CrossRef]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [Green Version]
- Wesolowski, J.; Alzogaray, V.; Reyelt, J.; Unger, M.; Juarez, K.; Urrutia, M.; Cauerhff, A.; Danquah, W.; Rissiek, B.; Scheuplein, F.; et al. Single domain antibodies: Promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009, 198, 157–174. [Google Scholar] [CrossRef] [Green Version]
- Conrath, K.E.; Wernery, U.; Muyldermans, S.; Nguyen, V.K. Emergence and evolution of functional heavy-chain antibodies in Camelidae. Dev. Comp. Immunol. 2003, 27, 87–103. [Google Scholar] [CrossRef]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Koch-Nolte, F. Perspectives for the Development of CD38-Specific Heavy Chain Antibodies as Therapeutics for Multiple Myeloma. Front. Immunol. 2018, 9, 2559. [Google Scholar] [CrossRef] [PubMed]
- Fumey, W.; Koenigsdorf, J.; Kunick, V.; Menzel, S.; Schutze, K.; Unger, M.; Schriewer, L.; Haag, F.; Adam, G.; Oberle, A.; et al. Nanobodies effectively modulate the enzymatic activity of CD38 and allow specific imaging of CD38(+) tumors in mouse models in vivo. Sci. Rep. 2017, 7, 14289. [Google Scholar] [CrossRef] [PubMed]
- Schriewer, L.; Schutze, K.; Petry, K.; Hambach, J.; Fumey, W.; Koenigsdorf, J.; Baum, N.; Menzel, S.; Rissiek, B.; Riecken, K.; et al. Nanobody-based CD38-specific heavy chain antibodies induce killing of multiple myeloma and other hematological malignancies. Theranostics 2019. (In press) [Google Scholar] [CrossRef]
- Schutze, K.; Petry, K.; Hambach, J.; Schuster, N.; Fumey, W.; Schriewer, L.; Rockendorf, J.; Menzel, S.; Albrecht, B.; Haag, F.; et al. CD38-Specific Biparatopic Heavy Chain Antibodies Display Potent Complement-Dependent Cytotoxicity Against Multiple Myeloma Cells. Front. Immunol. 2018, 9, 2553. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy - Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Weber, K.; Bartsch, U.; Stocking, C.; Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef]
- Weber, K.; Mock, U.; Petrowitz, B.; Bartsch, U.; Fehse, B. Lentiviral gene ontology (LeGO) vectors equipped with novel drug-selectable fluorescent proteins: New building blocks for cell marking and multi-gene analysis. Gene Ther. 2010, 17, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Koch-Nolte, F.; Reyelt, J.; Schossow, B.; Schwarz, N.; Scheuplein, F.; Rothenburg, S.; Haag, F.; Alzogaray, V.; Cauerhff, A.; Goldbaum, F.A. Single domain antibodies from llama effectively and specifically block T cell ecto-ADP-ribosyltransferase ART2.2 in vivo. FASEB J. 2007, 21, 3490–3498. [Google Scholar] [CrossRef]
- Schambach, A.; Mueller, D.; Galla, M.; Verstegen, M.M.; Wagemaker, G.; Loew, R.; Baum, C.; Bohne, J. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006, 13, 1524–1533. [Google Scholar] [CrossRef] [Green Version]
- Schambach, A.; Bohne, J.; Chandra, S.; Will, E.; Margison, G.P.; Williams, D.A.; Baum, C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol. Ther. 2006, 13, 391–400. [Google Scholar] [CrossRef]
- Frigyesi, I.; Adolfsson, J.; Ali, M.; Christophersen, M.K.; Johnsson, E.; Turesson, I.; Gullberg, U.; Hansson, M.; Nilsson, B. Robust isolation of malignant plasma cells in multiple myeloma. Blood 2014, 123, 1336–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK cell therapies at the forefront of cancer control. J. Clin. Invest. 2019, 129, 3499–3510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- An, N.; Hou, Y.N.; Zhang, Q.X.; Li, T.; Zhang, Q.L.; Fang, C.; Chen, H.; Lee, H.C.; Zhao, Y.J.; Du, X. Anti-Multiple Myeloma Activity of Nanobody-Based Anti-CD38 Chimeric Antigen Receptor T Cells. Mol. Pharm. 2018, 15, 4577–4588. [Google Scholar] [CrossRef]
- Bakhtiari, S.H.; Rahbarizadeh, F.; Hasannia, S.; Ahmadvand, D.; Iri-Sofla, F.J.; Rasaee, M.J. Anti-MUC1 nanobody can redirect T-body cytotoxic effector function. Hybrid 2009, 28, 85–92. [Google Scholar] [CrossRef]
- Hassani, M.; Hajari Taheri, F.; Sharifzadeh, Z.; Arashkia, A.; Hadjati, J.; van Weerden, W.M.; Modarressi, M.H.; Abolhassani, M. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J. Cell Biochem. 2019, 120, 10787–10795. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Groen, R.W.; Noort, W.A.; Themeli, M.; Lammerts van Bueren, J.J.; Parren, P.W.; Kuball, J.; Sebestyen, Z.; Yuan, H.; de Bruijn, J.; et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016, 101, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihara, K.; Yanagihara, K.; Takigahira, M.; Imai, C.; Kitanaka, A.; Takihara, Y.; Kimura, A. Activated T-cell-mediated immunotherapy with a chimeric receptor against CD38 in B-cell non-Hodgkin lymphoma. J. Immunother. 2009, 32, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Perna, F.; Berman, S.H.; Soni, R.K.; Mansilla-Soto, J.; Eyquem, J.; Hamieh, M.; Hendrickson, R.C.; Brennan, C.W.; Sadelain, M. Integrating Proteomics and Transcriptomics for Systematic Combinatorial Chimeric Antigen Receptor Therapy of AML. Cancer Cell 2017, 32, 506–519. [Google Scholar] [CrossRef] [Green Version]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody Based Dual Specific CARs. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hambach, J.; Riecken, K.; Cichutek, S.; Schütze, K.; Albrecht, B.; Petry, K.; Röckendorf, J.L.; Baum, N.; Kröger, N.; Hansen, T.; et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells 2020, 9, 321. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020321
Hambach J, Riecken K, Cichutek S, Schütze K, Albrecht B, Petry K, Röckendorf JL, Baum N, Kröger N, Hansen T, et al. Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs). Cells. 2020; 9(2):321. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020321
Chicago/Turabian StyleHambach, Julia, Kristoffer Riecken, Sophia Cichutek, Kerstin Schütze, Birte Albrecht, Katharina Petry, Jana Larissa Röckendorf, Natalie Baum, Nicolaus Kröger, Timon Hansen, and et al. 2020. "Targeting CD38-Expressing Multiple Myeloma and Burkitt Lymphoma Cells In Vitro with Nanobody-Based Chimeric Antigen Receptors (Nb-CARs)" Cells 9, no. 2: 321. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020321