Regulation of MT1-MMP Activity through Its Association with ERMs

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Cell Transfection
2.2. Antibodies and Reagents
2.3. Constructs and Reagents
2.4. Enzyme-Linked Immunosorbent Assay (ELISA) In Vitro Binding Assay
2.5. Co-Immunoprecipitation, Internalization and Immunoblot Assays
2.6. Fluorescence Confocal Microscopy, Fluorescent Lifetime Imaging-Förster Energy Transfer (FLIM-FRET) and Flow Cytometry
2.7. Exosome Isolation and Quantification
2.8. Extracellular Matrix (ECM) Degradation Assays
2.9. Statistical Analyses
3. Results
3.1. MT1-MMP Interacts with ERM (Ezrin, Radixin, Moesin) Proteins through Basic Residues in Its Cytoplasmic Tail
3.2. Mutation of the ERM Binding Site of MT1-MMP Alters the Steady State Subcellular Distribution of the Protein
3.3. Effect of ERM Binding on MT1-MMP Distribution in Membrane Microdomains
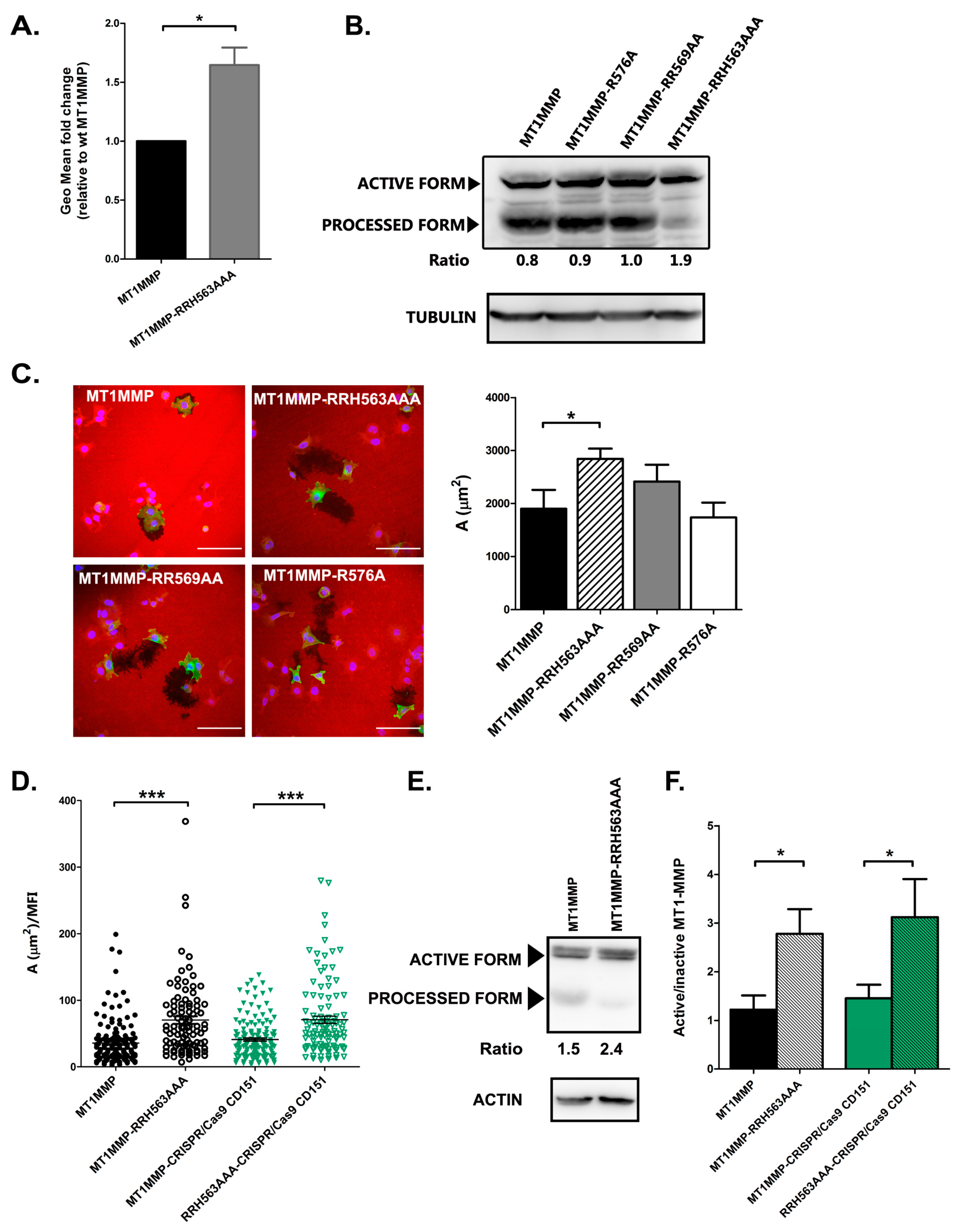
3.4. Effect of ERM Association on MT1-MMP Autoprocessing and Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Imai, K.; Ohuchi, E.; Aoki, T.; Nomura, H.; Fujii, Y.; Sato, H.; Seiki, M.; Okada, Y. Membrane-type matrix metalloproteinase 1 is a gelatinolytic enzyme and is secreted in a complex with tissue inhibitor of metalloproteinases 2. Cancer Res. 1996, 56, 2707–2710. [Google Scholar] [PubMed]
- Ohuchi, E.; Imai, K.; Fujii, Y.; Sato, H.; Seiki, M.; Okada, Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 1997, 272, 2446–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, Y.; Ito, N.; Nagase, H.; Evans, R.D.; Bird, S.A.; Seiki, M. Cell surface collagenolysis requires homodimerization of the membrane-bound collagenase MT1-MMP. Mol. Biol. Cell. 2006, 17, 5390–5399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauper, V.; Bailey, L.; Worley, J.R.; Soloway, P.; Patterson, M.L.; Murphy, G. Cellular activation of proMMP-13 by MT1-MMP depends on the C-terminal domain of MMP-13. FEBS Lett. 2002, 532, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef] [Green Version]
- Ratnikov, B.I.; Rozanov, D.V.; Postnova, T.I.; Baciu, P.G.; Zhang, H.; DiScipio, R.G.; Chestukhina, G.G.; Smith, J.W.; Deryugina, E.I.; Strongin, A.Y. An alternative processing of integrin alpha(v) subunit in tumor cells by membrane type-1 matrix metalloproteinase. J. Biol. Chem. 2002, 277, 7377–7385. [Google Scholar] [CrossRef] [Green Version]
- Belkin, A.M.; Akimov, S.S.; Zaritskaya, L.S.; Ratnikov, B.I.; Deryugina, E.I.; Strongin, A.Y. Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J. Biol. Chem. 2001, 276, 18415–18422. [Google Scholar] [CrossRef] [Green Version]
- Takino, T.; Koshikawa, N.; Miyamori, H.; Tanaka, M.; Sasaki, T.; Okada, Y.; Seiki, M.; Sato, H. Cleavage of metastasis suppressor gene product KiSS-1 protein/metastin by matrix metalloproteinases. Oncogene 2003, 22, 4617–4626. [Google Scholar] [CrossRef] [Green Version]
- Poincloux, R.; Lizarraga, F.; Chavrier, P. Matrix invasion by tumour cells: A focus on MT1-MMP trafficking to invadopodia. J. Cell. Sci. 2009, 122, 3015–3024. [Google Scholar] [CrossRef] [Green Version]
- Koziol, A.; Gonzalo, P.; Mota, A.; Pollan, A.; Lorenzo, C.; Colome, N.; Montaner, D.; Dopazo, J.; Arribas, J.; Canals, F.; et al. The protease MT1-MMP drives a combinatorial proteolytic program in activated endothelial cells. FASEB J. 2012, 26, 4481–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosse, C.; Lodillinsky, C.; Fuhrmann, L.; Nourieh, M.; Monteiro, P.; Irondelle, M.; Lagoutte, E.; Vacher, S.; Waharte, F.; Paul-Gilloteaux, P.; et al. Control of MT1-MMP transport by atypical PKC during breast-cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E1872–E1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Molina, J.; Gramolelli, S.; Liao, Z.; Carlson, J.W.; Ojala, P.M.; Lehti, K. MMP14 in Sarcoma: A Regulator of Tumor Microenvironment Communication in Connective Tissues. Cells 2019, 8, 991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, R.; Martin, G.; Tagit, O.; Guichard, A.; Cambi, A.; Voituriez, R.; Vassilopoulos, S.; Chavrier, P. MT1-MMP directs force-producing proteolytic contacts that drive tumor cell invasion. Nat. Commun. 2019, 10, 4886. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castro, A.; Marchesin, V.; Monteiro, P.; Lodillinsky, C.; Rosse, C.; Chavrier, P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell. Dev. Biol. 2016, 32, 555–576. [Google Scholar] [CrossRef]
- Gifford, V.; Itoh, Y. MT1-MMP-dependent cell migration: Proteolytic and non-proteolytic mechanisms. Biochem. Soc. Trans. 2019, 47, 811–826. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44–46, 207–223. [Google Scholar] [CrossRef]
- Itoh, Y.; Ito, N.; Nagase, H.; Seiki, M. The second dimer interface of MT1-MMP, the transmembrane domain, is essential for ProMMP-2 activation on the cell surface. J. Biol. Chem. 2008, 283, 13053–13062. [Google Scholar] [CrossRef] [Green Version]
- Lehti, K.; Lohi, J.; Juntunen, M.M.; Pei, D.; Keski-Oja, J. Oligomerization through hemopexin and cytoplasmic domains regulates the activity and turnover of membrane-type 1 matrix metalloproteinase. J. Biol. Chem. 2002, 277, 8440–8448. [Google Scholar] [CrossRef] [Green Version]
- Tam, E.M.; Wu, Y.I.; Butler, G.S.; Stack, M.S.; Overall, C.M. Collagen binding properties of the membrane type-1 matrix metalloproteinase (MT1-MMP) hemopexin C domain. The ectodomain of the 44-kDa autocatalytic product of MT1-MMP inhibits cell invasion by disrupting native type I collagen cleavage. J. Biol. Chem. 2002, 277, 39005–39014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.A.; Osenkowski, P.; Zhao, H.; Kim, S.; Toth, M.; Cole, K.; Aboukameel, A.; Saliganan, A.; Schuger, L.; Bonfil, R.D.; et al. The inactive 44-kDa processed form of membrane type 1 matrix metalloproteinase (MT1-MMP) enhances proteolytic activity via regulation of endocytosis of active MT1-MMP. J. Biol. Chem. 2008, 283, 17391–17405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Barrantes, S.; Toth, M.; Bernardo, M.M.; Yurkova, M.; Gervasi, D.C.; Raz, Y.; Sang, Q.A.; Fridman, R. Binding of active (57 kDa) membrane type 1-matrix metalloproteinase (MT1-MMP) to tissue inhibitor of metalloproteinase (TIMP)-2 regulates MT1-MMP processing and pro-MMP-2 activation. J. Biol. Chem. 2000, 275, 12080–12089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, A.; Hoshino, D.; Koshikawa, N.; Seiki, M.; Suzuki, T.; Ichikawa, K. Critical role of transient activity of MT1-MMP for ECM degradation in invadopodia. PLoS Comput. Biol. 2013, 9, e1003086. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdes, M.; Sanchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell. Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef]
- Schroder, H.M.; Hoffmann, S.C.; Hecker, M.; Korff, T.; Ludwig, T. The tetraspanin network modulates MT1-MMP cell surface trafficking. Int. J. Biochem. Cell. Biol. 2013, 45, 1133–1144. [Google Scholar] [CrossRef]
- Hong, I.K.; Byun, H.J.; Lee, J.; Jin, Y.J.; Wang, S.J.; Jeoung, D.I.; Kim, Y.M.; Lee, H. The tetraspanin CD81 protein increases melanoma cell motility by up-regulating metalloproteinase MT1-MMP expression through the pro-oncogenic Akt-dependent Sp1 activation signaling pathways. J. Biol. Chem. 2014, 289, 15691–15704. [Google Scholar] [CrossRef] [Green Version]
- Takino, T.; Miyamori, H.; Kawaguchi, N.; Uekita, T.; Seiki, M.; Sato, H. Tetraspanin CD63 promotes targeting and lysosomal proteolysis of membrane-type 1 matrix metalloproteinase. Biochem. Biophys. Res. Commun. 2003, 304, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Yanez-Mo, M.; Barreiro, O.; Gonzalo, P.; Batista, A.; Megias, D.; Genis, L.; Sachs, N.; Sala-Valdes, M.; Alonso, M.A.; Montoya, M.C.; et al. MT1-MMP collagenolytic activity is regulated through association with tetraspanin CD151 in primary endothelial cells. Blood 2008, 112, 3217–3226. [Google Scholar] [CrossRef]
- Lafleur, M.A.; Xu, D.; Hemler, M.E. Tetraspanin proteins regulate membrane type-1 matrix metalloproteinase-dependent pericellular proteolysis. Mol. Biol. Cell. 2009, 20, 2030–2040. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, C.; Roche, S.; Benistant, C. Vesicular trafficking regulators are new players in breast cancer progression: Role of TOM1L1 in ERBB2-dependent invasion. Mol. Cell. Oncol. 2016, 3, e1182241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, F.; He, J.; Wu, P.; Tay, L.W.R.; Cai, M.; Nian, W.; Weng, Y.; Qin, L.; Chang, J.T.; et al. Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev. Cell. 2017, 43, 186–197 e187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, T.; Hasegawa, K.; Aoki, Y.; Watanabe, T.; Otagiri, Y.; Arasaki, K.; Wakana, Y.; Asano, K.; Tanaka, M.; Yamaguchi, H.; et al. MT1-MMP recruits the ER-Golgi SNARE Bet1 for efficient MT1-MMP transport to the plasma membrane. J. Cell Biol. 2019, 218, 3355–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.C.; Coppolino, M.G. Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. J. Biol. Chem. 2011, 286, 43405–43416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyalendo, C.; Michaud, M.; Beaulieu, E.; Roghi, C.; Murphy, G.; Gingras, D.; Beliveau, R. Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: Role in endothelial and tumor cell migration. J. Biol. Chem. 2007, 282, 15690–15699. [Google Scholar] [CrossRef] [Green Version]
- Remacle, A.; Murphy, G.; Roghi, C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J. Cell. Sci. 2003, 116, 3905–3916. [Google Scholar] [CrossRef] [Green Version]
- Galvez, B.G.; Matias-Roman, S.; Yanez-Mo, M.; Sanchez-Madrid, F.; Arroyo, A.G. ECM regulates MT1-MMP localization with beta1 or alphavbeta3 integrins at distinct cell compartments modulating its internalization and activity on human endothelial cells. J. Cell. Biol. 2002, 159, 509–521. [Google Scholar] [CrossRef]
- Lagoutte, E.; Villeneuve, C.; Lafanechere, L.; Wells, C.M.; Jones, G.E.; Chavrier, P.; Rosse, C. LIMK Regulates Tumor-Cell Invasion and Matrix Degradation Through Tyrosine Phosphorylation of MT1-MMP. Sci. Rep. 2016, 6, 24925. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Marrero-Diaz, R.; Megias, D.; Genis, L.; Garcia-Grande, A.; Garcia, M.A.; Arroyo, A.G.; Montoya, M.C. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J. 2007, 26, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Steffen, A.; Le Dez, G.; Poincloux, R.; Recchi, C.; Nassoy, P.; Rottner, K.; Galli, T.; Chavrier, P. MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr. Biol. 2008, 18, 926–931. [Google Scholar] [CrossRef]
- Planchon, D.; Rios Morris, E.; Genest, M.; Comunale, F.; Vacher, S.; Bieche, I.; Denisov, E.V.; Tashireva, L.A.; Perelmuter, V.M.; Linder, S.; et al. MT1-MMP targeting to endolysosomes is mediated by upregulation of flotillins. J. Cell. Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Galvez, B.G.; Matias-Roman, S.; Yanez-Mo, M.; Vicente-Manzanares, M.; Sanchez-Madrid, F.; Arroyo, A.G. Caveolae are a novel pathway for membrane-type 1 matrix metalloproteinase traffic in human endothelial cells. Mol. Biol. Cell. 2004, 15, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, D.; Kirkbride, K.C.; Costello, K.; Clark, E.S.; Sinha, S.; Grega-Larson, N.; Tyska, M.J.; Weaver, A.M. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell. Rep. 2013, 5, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakulinen, J.; Sankkila, L.; Sugiyama, N.; Lehti, K.; Keski-Oja, J. Secretion of active membrane type 1 matrix metalloproteinase (MMP-14) into extracellular space in microvesicular exosomes. J. Cell. Biochem. 2008, 105, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Cao, H.; Chen, J.; Weller, S.G.; Krueger, E.W.; Zhang, L.; Razidlo, G.L.; McNiven, M.A. Pancreatic tumor cell metastasis is restricted by MT1-MMP binding protein MTCBP-1. J. Cell Biol. 2019, 218, 317–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anilkumar, N.; Uekita, T.; Couchman, J.R.; Nagase, H.; Seiki, M.; Itoh, Y. Palmitoylation at Cys574 is essential for MT1-MMP to promote cell migration. FASEB J. 2005, 19, 1326–1328. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, P.; Guadamillas, M.C.; Hernandez-Riquer, M.V.; Pollan, A.; Grande-Garcia, A.; Bartolome, R.A.; Vasanji, A.; Ambrogio, C.; Chiarle, R.; Teixido, J.; et al. MT1-MMP is required for myeloid cell fusion via regulation of Rac1 signaling. Dev. Cell 2010, 18, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, T.; Seiki, M. A membrane protease regulates energy production in macrophages by activating hypoxia-inducible factor-1 via a non-proteolytic mechanism. J. Biol. Chem. 2010, 285, 29951–29964. [Google Scholar] [CrossRef] [Green Version]
- Terawaki, S.; Kitano, K.; Aoyama, M.; Mori, T.; Hakoshima, T. MT1-MMP recognition by ERM proteins and its implication in CD44 shedding. Genes Cells 2015, 20, 847–859. [Google Scholar] [CrossRef] [Green Version]
- McClatchey, A.I. ERM proteins at a glance. J. Cell. Sci. 2014, 127, 3199–3204. [Google Scholar] [CrossRef] [Green Version]
- Ponuwei, G.A. A glimpse of the ERM proteins. J. Biomed. Sci. 2016, 23, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukita, S.; Oishi, K.; Sato, N.; Sagara, J.; Kawai, A.; Tsukita, S. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J. Cell Biol. 1994, 126, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Serrador, J.M.; Alonso-Lebrero, J.L.; del Pozo, M.A.; Furthmayr, H.; Schwartz-Albiez, R.; Calvo, J.; Lozano, F.; Sanchez-Madrid, F. Moesin interacts with the cytoplasmic region of intercellular adhesion molecule-3 and is redistributed to the uropod of T lymphocytes during cell polarization. J. Cell Biol. 1997, 138, 1409–1423. [Google Scholar] [CrossRef] [Green Version]
- Sala-Valdes, M.; Ursa, A.; Charrin, S.; Rubinstein, E.; Hemler, M.E.; Sanchez-Madrid, F.; Yanez-Mo, M. EWI-2 and EWI-F link the tetraspanin web to the actin cytoskeleton through their direct association with ezrin-radixin-moesin proteins. J. Biol. Chem. 2006, 281, 19665–19675. [Google Scholar] [CrossRef] [Green Version]
- Fehon, R.G.; McClatchey, A.I.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell. Biol. 2010, 11, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Zhou, C.X.; Gao, Y. Moesin regulates the motility of oral cancer cells via MT1-MMP and E-cadherin/p120-catenin adhesion complex. Oral. Oncol. 2015, 51, 935–943. [Google Scholar] [CrossRef]
- Galvez, B.G.; Matias-Roman, S.; Albar, J.P.; Sanchez-Madrid, F.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J. Biol. Chem. 2001, 276, 37491–37500. [Google Scholar] [CrossRef] [Green Version]
- Yanez-Mo, M.; Alfranca, A.; Cabanas, C.; Marazuela, M.; Tejedor, R.; Ursa, M.A.; Ashman, L.K.; de Landazuri, M.O.; Sanchez-Madrid, F. Regulation of endothelial cell motility by complexes of tetraspan molecules CD81/TAPA-1 and CD151/PETA-3 with alpha3 beta1 integrin localized at endothelial lateral junctions. J. Cell Biol. 1998, 141, 791–804. [Google Scholar] [CrossRef]
- Rocha-Perugini, V.; Gonzalez-Granado, J.M.; Tejera, E.; Lopez-Martin, S.; Yanez-Mo, M.; Sanchez-Madrid, F. Tetraspanins CD9 and CD151 at the immune synapse support T-cell integrin signaling. Eur. J. Immunol. 2014, 44, 1967–1975. [Google Scholar] [CrossRef] [Green Version]
- Barreiro, O.; Yanez-Mo, M.; Serrador, J.M.; Montoya, M.C.; Vicente-Manzanares, M.; Tejedor, R.; Furthmayr, H.; Sanchez-Madrid, F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J. Cell Biol. 2002, 157, 1233–1245. [Google Scholar] [CrossRef] [Green Version]
- Caiolfa, V.R.; Zamai, M.; Malengo, G.; Andolfo, A.; Madsen, C.D.; Sutin, J.; Digman, M.A.; Gratton, E.; Blasi, F.; Sidenius, N. Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies. J. Cell Biol. 2007, 179, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Caiolfa, V.R.; Zamai, M.; Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008, 94, L14–L16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Suarez, H.; Gamez-Valero, A.; Reyes, R.; Lopez-Martin, S.; Rodriguez, M.J.; Carrascosa, J.L.; Cabanas, C.; Borras, F.E.; Yanez-Mo, M. A bead-assisted flow cytometry method for the semi-quantitative analysis of Extracellular Vesicles. Sci. Rep. 2017, 7, 11271. [Google Scholar] [CrossRef] [Green Version]
- Campos-Silva, C.; Suarez, H.; Jara-Acevedo, R.; Linares-Espinos, E.; Martinez-Pineiro, L.; Yanez-Mo, M.; Vales-Gomez, M. High sensitivity detection of extracellular vesicles immune-captured from urine by conventional flow cytometry. Sci. Rep. 2019, 9, 2042. [Google Scholar] [CrossRef] [Green Version]
- Baldassarre, M.; Pompeo, A.; Beznoussenko, G.; Castaldi, C.; Cortellino, S.; McNiven, M.A.; Luini, A.; Buccione, R. Dynamin participates in focal extracellular matrix degradation by invasive cells. Mol Biol Cell 2003, 14, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Yonemura, S.; Hirao, M.; Doi, Y.; Takahashi, N.; Kondo, T.; Tsukita, S.; Tsukita, S. Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J. Cell Biol. 1998, 140, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Baker, T.M.; Waheed, S.; Syed, V. RNA interference screening identifies clathrin-B and cofilin-1 as mediators of MT1-MMP in endometrial cancer. Exp Cell Res. 2018, 370, 663–670. [Google Scholar] [CrossRef]
- Brasher, M.I.; Martynowicz, D.M.; Grafinger, O.R.; Hucik, A.; Shanks-Skinner, E.; Uniacke, J.; Coppolino, M.G. Interaction of Munc18c and syntaxin4 facilitates invadopodium formation and extracellular matrix invasion of tumor cells. J. Biol. Chem. 2017, 292, 16199–16210. [Google Scholar] [CrossRef] [Green Version]
- Miyata, T.; Ohnishi, H.; Suzuki, J.; Yoshikumi, Y.; Ohno, H.; Mashima, H.; Yasuda, H.; Ishijima, T.; Osawa, H.; Satoh, K.; et al. Involvement of syntaxin 4 in the transport of membrane-type 1 matrix metalloproteinase to the plasma membrane in human gastric epithelial cells. Biochem Biophys Res. Commun. 2004, 323, 118–124. [Google Scholar] [CrossRef]
- Monteiro, P.; Rosse, C.; Castro-Castro, A.; Irondelle, M.; Lagoutte, E.; Paul-Gilloteaux, P.; Desnos, C.; Formstecher, E.; Darchen, F.; Perrais, D.; et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J. Cell Biol. 2013, 203, 1063–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvez, B.G.; Genis, L.; Matias-Roman, S.; Oblander, S.A.; Tryggvason, K.; Apte, S.S.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J. Biol. Chem. 2005, 280, 1292–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, S.; Koch, M.; Nubel, T.; Ladwein, M.; Antolovic, D.; Klingbeil, P.; Hildebrand, D.; Moldenhauer, G.; Langbein, L.; Franke, W.W.; et al. A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol. Cancer Res. 2007, 5, 553–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrero-Diaz, R.; Bravo-Cordero, J.J.; Megias, D.; Garcia, M.A.; Bartolome, R.A.; Teixido, J.; Montoya, M.C. Polarized MT1-MMP-CD44 interaction and CD44 cleavage during cell retraction reveal an essential role for MT1-MMP in CD44-mediated invasion. Cell. Motil. Cytoskelet. 2009, 66, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Zhao, L.; Wu, H.; Zhao, H.; Yu, Z.; He, M.; Jin, F.; Wei, M. Moesin is an independent prognostic marker for ER-positive breast cancer. Oncol. Lett. 2019, 17, 1921–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez, H.; López-Martín, S.; Toribio, V.; Zamai, M.; Hernández-Riquer, M.V.; Genís, L.; Arroyo, A.G.; Yáñez-Mó, M. Regulation of MT1-MMP Activity through Its Association with ERMs. Cells 2020, 9, 348. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020348
Suárez H, López-Martín S, Toribio V, Zamai M, Hernández-Riquer MV, Genís L, Arroyo AG, Yáñez-Mó M. Regulation of MT1-MMP Activity through Its Association with ERMs. Cells. 2020; 9(2):348. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020348
Chicago/Turabian StyleSuárez, Henar, Soraya López-Martín, Víctor Toribio, Moreno Zamai, M. Victoria Hernández-Riquer, Laura Genís, Alicia G. Arroyo, and María Yáñez-Mó. 2020. "Regulation of MT1-MMP Activity through Its Association with ERMs" Cells 9, no. 2: 348. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9020348