Characterising the Transcriptional and Translational Impact of the Schizophrenia-Associated miR-1271-5p in Neuronal Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Neuronal Differentiation
2.3. miR-1271-5p Transfections
2.4. RT-qPCR Quantification of miRNA Expression
2.5. Ribosome Profiling
2.6. mRNA Sequencing
2.7. Processing of Sequencing Data
2.8. Differential Expression Analysis
2.9. Functional Analysis
2.10. Analysis of miR-1271-5p Target Genes
3. Results
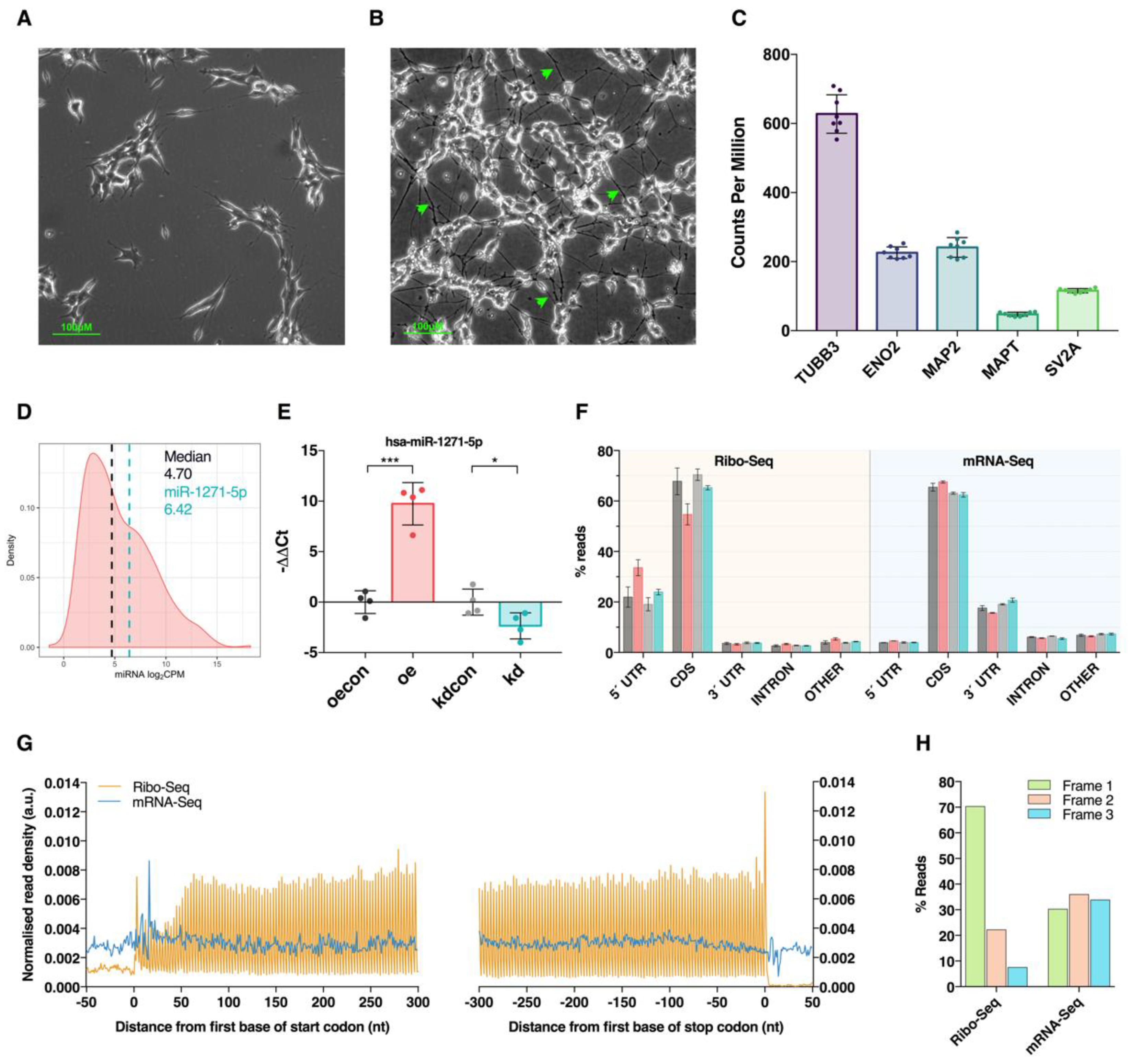
3.1. Modulation of miR-1271-5p Expression In Vitro
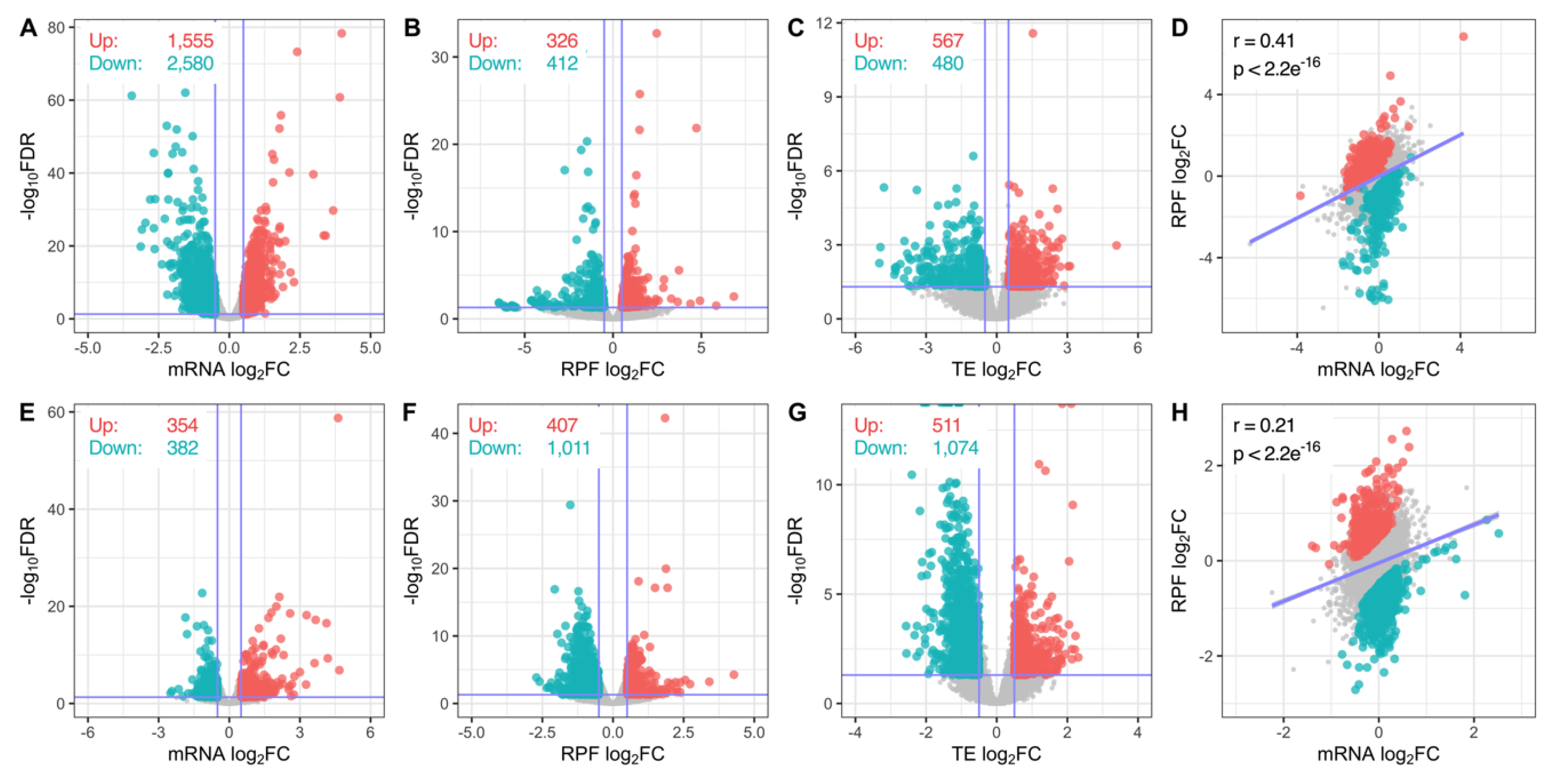
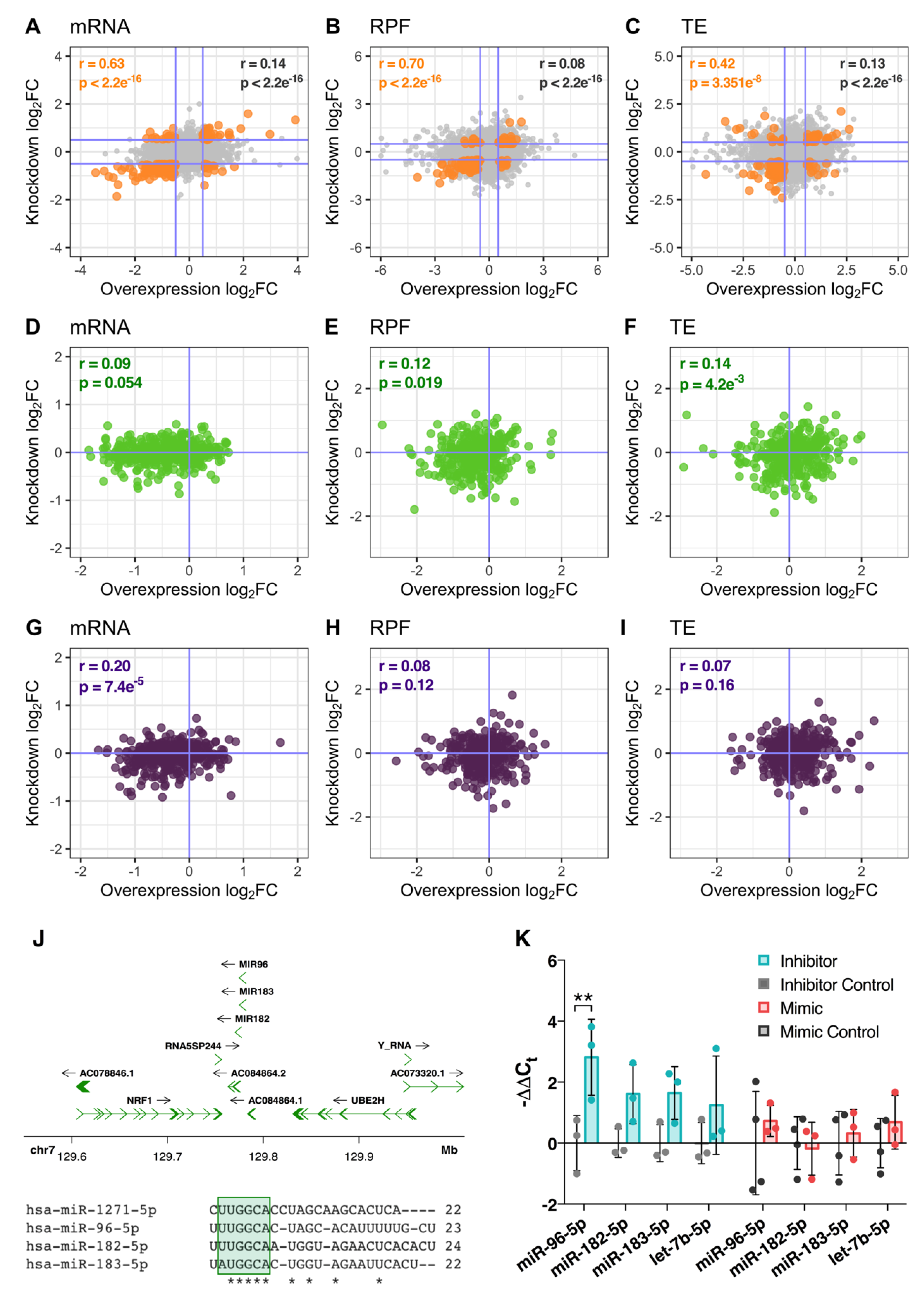
3.2. Transcriptome-Wide Remodelling Induced by miR-1271-5p Differential Expression
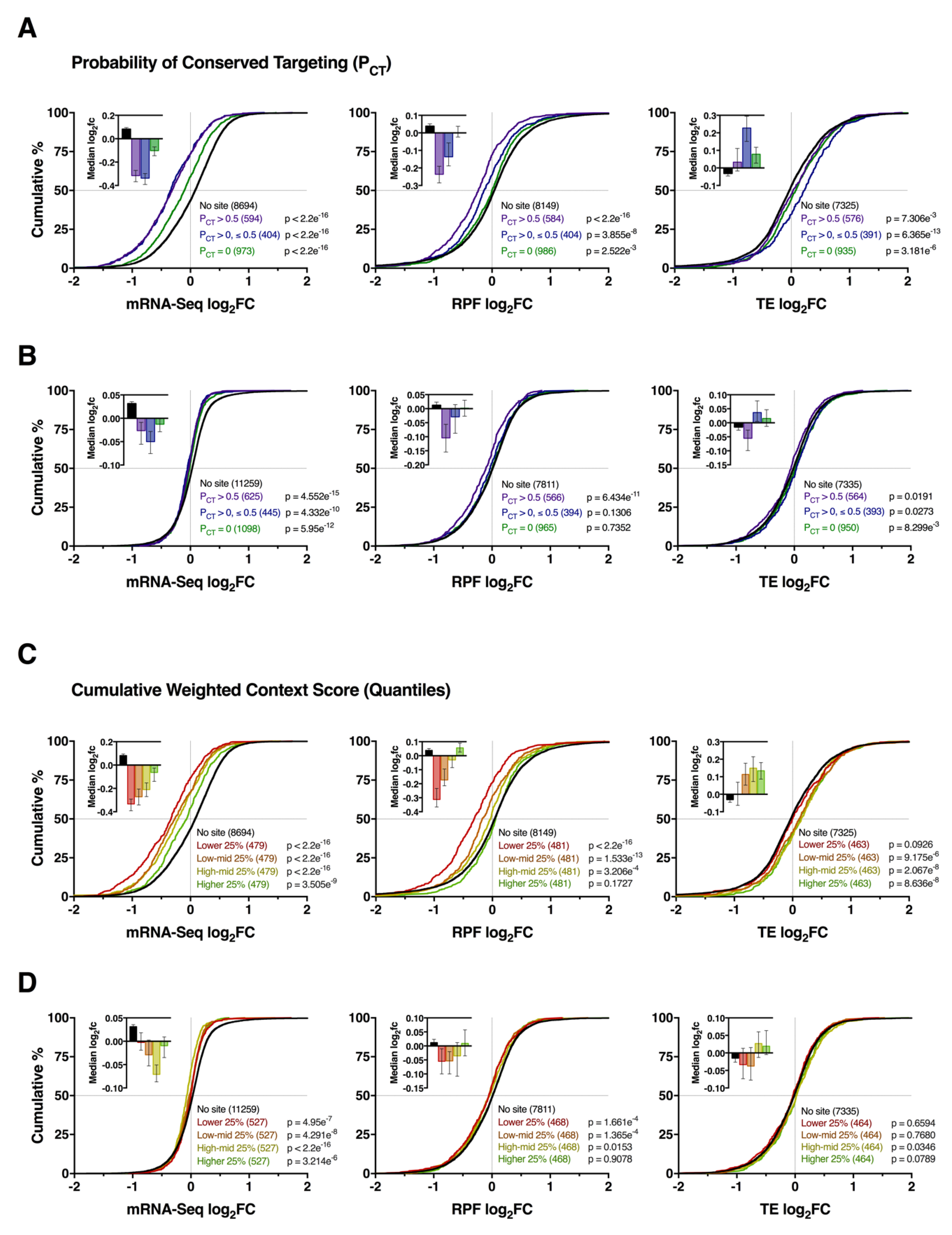
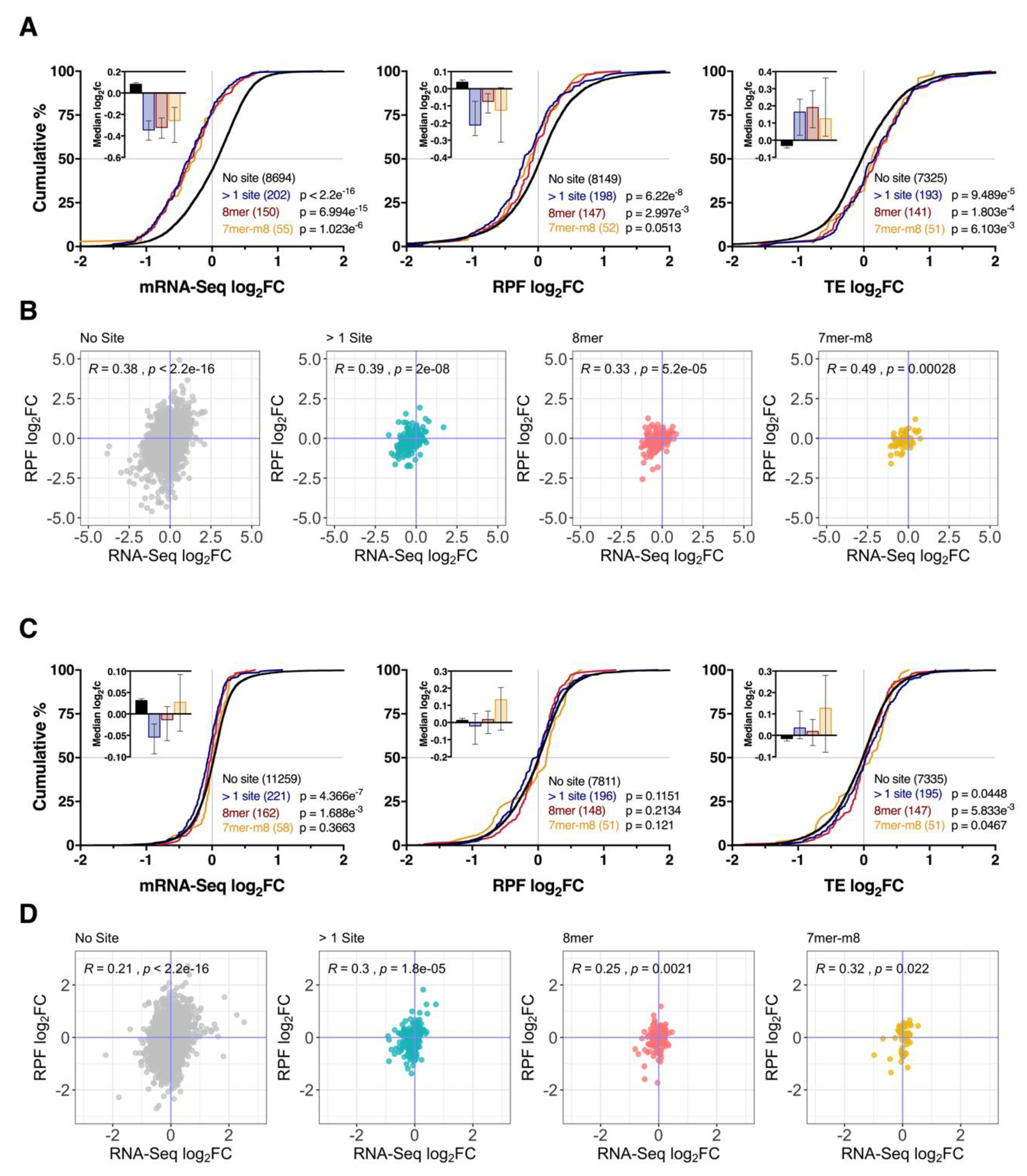
3.3. Impact of Conserved Targeting and Binding Site Context on mRNA Repression
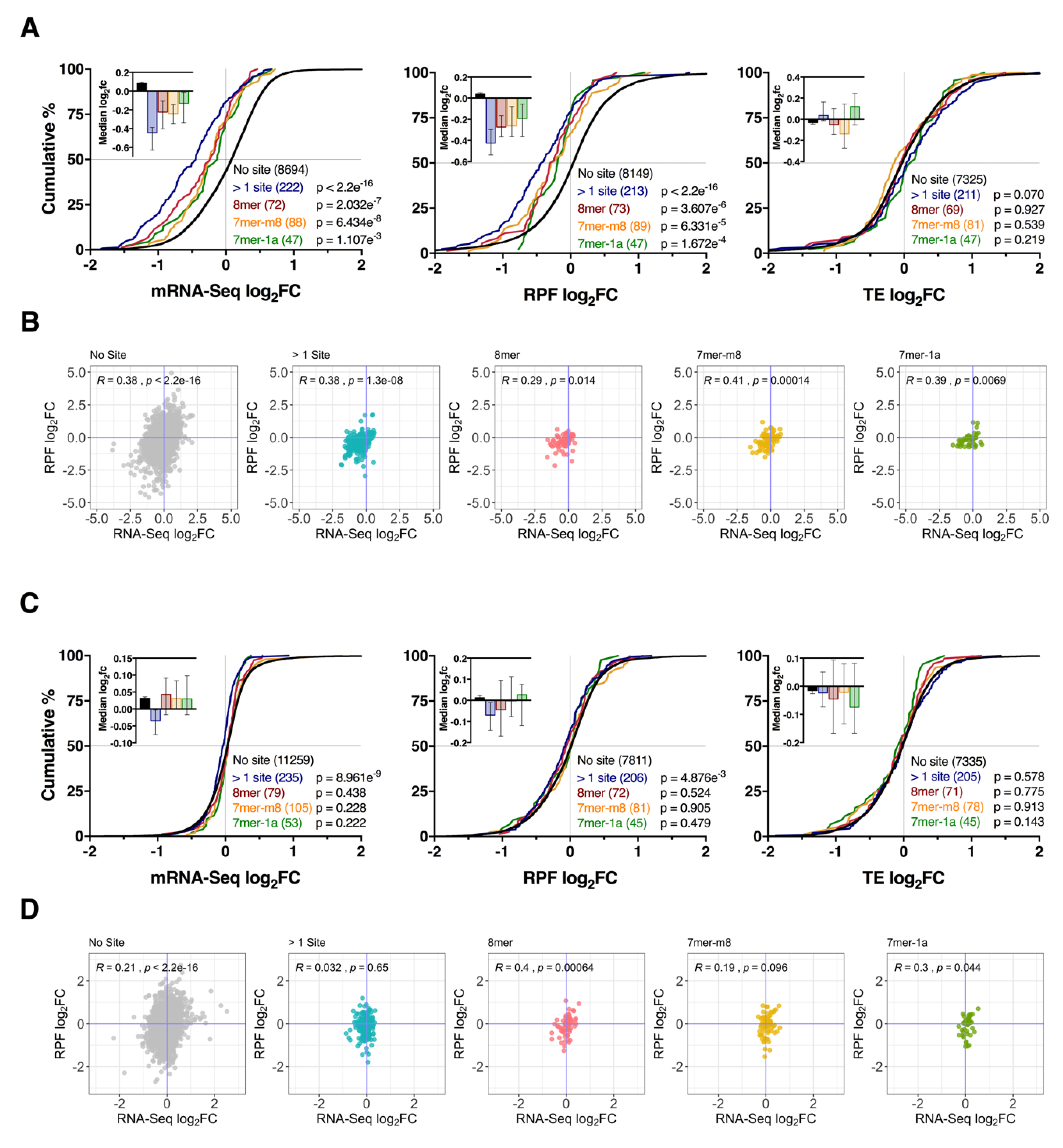
3.4. Effect of Seed Complementarity on Target mRNA Expression
3.5. Repression of mRNA via Binding Sites within the Coding Sequence
3.6. miR-1271-5p Knockdown Triggers Upregulation of miRNAs with Similar Seed Sequence
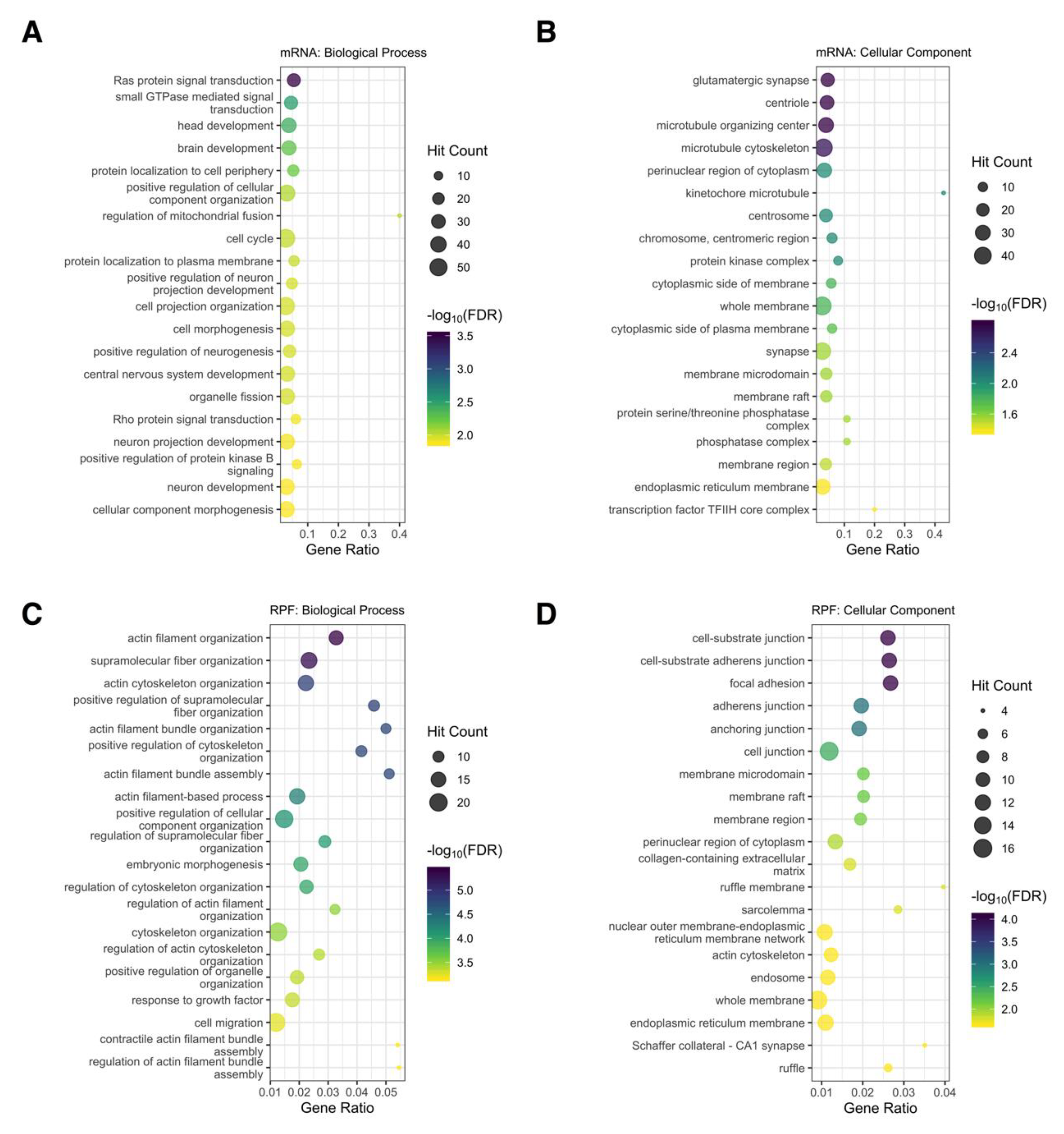
3.7. Functional Analysis of Differentially Regulated Genes
3.8. Functional Analysis of Genes Directly Targeted by miR-1271-5p
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Natera-Naranjo, O.; Aschrafi, A.; Gioio, A.E.; Kaplan, B.B. Identification and quantitative analyses of microRNAs located in the distal axons of sympathetic neurons. RNA 2010, 16, 1516–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Chen, Q.; Zen, K.; Zhang, C.Y.; Zhang, Q. Synaptosomes secrete and uptake functionally active microRNAs via exocytosis and endocytosis pathways. J. Neurochem. 2012, 124, 15–25. [Google Scholar] [CrossRef]
- Sambandan, S.; Akbalik, G.; Kochen, L.; Rinne, J.; Kahlstatt, J.; Glock, C.; Tushev, G.; Alvarez-Castelao, B.; Heckel, A.; Schuman, E.M. Activity-dependent spatially localized miRNA maturation in neuronal dendrites. Science 2017, 355, 634–637. [Google Scholar] [CrossRef]
- Lugli, G.; Larson, J.; Martone, M.E.; Jones, Y.; Smalheiser, N.R. Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J. Neurochem. 2005, 94, 896–905. [Google Scholar] [CrossRef]
- Eacker, S.M.; Keuss, M.J.; Berezikov, E.; Dawson, V.L.; Dawson, T.M. Neuronal Activity Regulates Hippocampal miRNA Expression. PLoS ONE 2011, 6, e25068. [Google Scholar] [CrossRef] [Green Version]
- Schratt, G.M.; Tuebing, F.; Nigh, E.A.; Kane, C.G.; Sabatini, M.E.; Kiebler, M.; Greenberg, M.E. A brain-specific microRNA regulates dendritic spine development. Nature 2006, 439, 283–289. [Google Scholar] [CrossRef]
- Neo, W.H.; Yap, K.; Lee, S.H.; Looi, L.S.; Khandelia, P.; Neo, S.X.; Makeyev, E.V.; Su, I.-H. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning Ezh2 expression. J. Boil. Chem. 2014, 289, 20788–20801. [Google Scholar] [CrossRef] [Green Version]
- Siegert, S.; Seo, J.; Kwon, E.J.; Rudenko, A.; Cho, S.; Wang, W.; Flood, Z.; Martorell, A.J.; Ericsson, M.; Mungenast, A.E.; et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nat. Neurosci. 2015, 18, 1008–1016. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Song, J.; Ouyang, Y.; Han, Q.; Chen, W.; Zhao, X.; Xie, Y.; Chen, Y.; Yuan, W.-E.; Fan, C. Advances in Roles of miR-132 in the Nervous System. Front. Pharmacol. 2017, 8, 770. [Google Scholar] [CrossRef]
- Kiltschewskij, D.; Cairns, M.J. Temporospatial guidance of activity-dependent gene expression by microRNA: Mechanisms and functional implications for neural plasticity. Nucleic Acids Res. 2018, 47, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, B. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Geaghan, M.; Cairns, M.J. MicroRNA and Posttranscriptional Dysregulation in Psychiatry. Boil. Psychiatry 2015, 78, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issler, O.; Chen, A. Determining the role of microRNAs in psychiatric disorders. Nat. Rev. Neurosci. 2015, 16, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.P.; Covault, J. Human miR-1271 is a miR-96 paralog with distinct non-conserved brain expression pattern. Nucleic Acids Res. 2010, 39, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Geaghan, M.P.; Atkins, J.R.; Brichta, A.M.; Tooney, P.A.; Scott, R.J.; Carr, V.J.; Cairns, M.J. Alteration of miRNA-mRNA interactions in lymphocytes of individuals with schizophrenia. J. Psychiatr. Res. 2019, 112, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.-C.; Wu, J.; Zhang, L.; Zhang, G.-L.; Sui, J.; Tong, W.-W.; Zhang, X.-Y.; Nie, L.-L.; Duan, J.-H.; Zhang, L.-R.; et al. Alterations of miR-132 are novel diagnostic biomarkers in peripheral blood of schizophrenia patients. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2015, 63, 23–29. [Google Scholar] [CrossRef]
- Balcells, I.; Cirera, S.; Busk, P.K. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 2011, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Boil. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.G.; Weissman, J.S. Plastid: Nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genom. 2016, 17, 958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Karaletsos, T.; Drewe, P.; Sreedharan, V.T.; Kuo, D.; Singh, K.; Wendel, H.-G.; Rätsch, G. RiboDiff: Detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics 2016, 33, 139–141. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, B. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Boil. 2011, 18, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Mockly, S.; Seitz, H. Inconsistencies and Limitations of Current MicroRNA Target Identification Methods. Breast Cancer 2019, 1970, 291–314. [Google Scholar] [CrossRef]
- Friedman, R.; Farh, K.K.-H.; Burge, C.B.; Bartel, B. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008, 19, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Zhang, X.; Cai, Z.; Zhou, J.; Cao, R.; Zhao, Y.; Chen, Z.; Wang, D.; Ruan, W.; Zhao, Q.; et al. A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Boil. 2018, 25, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Syed, A.P.; Bilen, B.; Zavolan, M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013, 23, 604–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brümmer, A.; Hausser, J. MicroRNA binding sites in the coding region of mRNAs: Extending the repertoire of post-transcriptional gene regulation. BioEssays 2014, 36, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bao, J.; Kim, M.; Yuan, S.; Tang, C.; Zheng, H.; Mastick, G.; Xu, C.; Yan, W. Two miRNA clusters, miR-34b/c and miR-449, are essential for normal brain development, motile ciliogenesis, and spermatogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, E2851–E2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, J.; Li, D.; Wang, L.; Wu, J.; Hu, Y.; Wang, Z.; Chen, Y.; Cao, X.; Jiang, C.; Yan, W.; et al. MicroRNA-449 and MicroRNA-34b/c Function Redundantly in Murine Testes by Targeting E2F Transcription Factor-Retinoblastoma Protein (E2F-pRb) Pathway. J. Boil. Chem. 2012, 287, 21686–21698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-F.; Yang, Y.-F.; Peng, Z.-P.; Zhang, M.-F.; Liang, J.-Y.; Chen, W.; Liu, X.-H.; Zheng, Y.-L. FOXK2, regulted by miR-1271-5p, promotes cell growth and indicates unfavorable prognosis in hepatocellular carcinoma. Int. J. Biochem. Cell Boil. 2017, 88, 155–161. [Google Scholar] [CrossRef]
- Chen, X.; Yang, S.; Zeng, J.; Chen, M. miR-1271-5p inhibits cell proliferation and induces apoptosis in acute myeloid leukemia by targeting ZIC2. Mol. Med. Rep. 2018, 19, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villen, J.; Bartel, B. mRNA destabilization is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Ingolia, N.T.; Weissman, J.S.; Bartel, B. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010, 466, 835–840. [Google Scholar] [CrossRef] [Green Version]
- Bazzini, A.A.; Lee, M.T.; Giraldez, A.J. Ribosome Profiling Shows That miR-430 Reduces Translation Before Causing mRNA Decay in Zebrafish. Science 2012, 336, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, B.P.; Burge, C.B.; Bartel, B. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnall-Levin, M.; Rissland, O.S.; Johnston, W.K.; Perrimon, N.; Bartel, B.; Berger, B. Unusually effective microRNA targeting within repeat-rich coding regions of mammalian mRNAs. Genome Res. 2011, 21, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guojing, L.; Zhang, R.; Xu, J.; Wu, C.-I.; Lu, X. Functional Conservation of Both CDS- and 3′-UTR-Located MicroRNA Binding Sites between Species. Mol. Boil. Evol. 2015, 32, 3276. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.E.; Grünhagen, J.; Jäger, M.; Horbelt, D.; Schwill, S.; Kallenbach, K.; Guo, G.; Manke, T.; Knaus, P.; Mundlos, S.; et al. MicroRNAs Differentially Expressed in Postnatal Aortic Development Downregulate Elastin via 3′ UTR and Coding-Sequence Binding Sites. PLoS ONE 2011, 6, e16250. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lin, A.; Hung, Y.; Chen, K.-D.; Wu, C.; Lie, C.; Hsiao, C.-C.; Chen, C.-J.; Liu, S.; Fang, W.-F.; et al. Whole genome gene expression changes and hematological effects of rikkunshito in patients with advanced non-small cell lung cancer receiving first line chemotherapy. Exp. Ther. Med. 2017, 14, 2040–2052. [Google Scholar] [CrossRef] [Green Version]
- Fikri, R.M.N.; Norlelawati, A.T.; El-Huda, A.R.N.; Hanisah, M.N.; Kartini, A.; Norsidah, K.; Zamzila, A.N. Reelin (RELN) DNA methylation in the peripheral blood of schizophrenia. J. Psychiatr. Res. 2017, 88, 28–37. [Google Scholar] [CrossRef]
- Gessert, S.; Bugner, V.; Tecza, A.; Pinker, M.; Kühl, M. FMR1/FXR1 and the miRNA pathway are required for eye and neural crest development. Dev. Biol. 2010, 341, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Schlüter, T.; Berger, C.; Rosengauer, E.; Fieth, P.; Krohs, C.; Ushakov, K.; Steel, K.P.; Avraham, K.B.; Hartmann, A.K.; Felmy, F.; et al. miR-96 is required for normal development of the auditory hindbrain. Hum. Mol. Genet. 2018, 27, 860–874. [Google Scholar] [CrossRef]
- Davari, M.; Soheili, Z.S.; Samiei, S.; Sharifi, Z.; Pirmardan, E.R. Overexpression of miR-183/-96/-182 triggers neuronal cell fate in Human Retinal Pigment Epithelial (hRPE) cells in culture. Biochem. Biophys. Res. Commun. 2017, 483, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.-W.; Ma, L.; Phillips, C.; Zhang, S.-C. miR-200 and miR-96 families repress neural induction from human embryonic stem cells. Development 2013, 140, 2611–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Jia, L.; Li, Y.; Ebrahim, S.; May-Simera, H.; Wood, A.; Morell, R.J.; Liu, P.; Lei, J.; Kachar, B.; et al. Maturation arrest in early postnatal sensory receptors by deletion of the miR-183/96/182 cluster in mouse. Proc. Natl. Acad. Sci. USA 2017, 114, E4271–E4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woldemichael, B.T.; Jawaid, A.; Kremer, E.A.; Gaur, N.; Krol, J.; Marchais, A.; Mansuy, I.M. The microRNA cluster miR-183/96/182 contributes to long-term memory in a protein phosphatase 1-dependent manner. Nat. Commun. 2016, 7, 12594. [Google Scholar] [CrossRef] [Green Version]
- Jawaid, A.; Woldemichael, B.T.; Kremer, E.A.; Laferriere, F.; Gaur, N.; Afroz, T.; Polymenidou, M.; Mansuy, I.M. Memory Decline and Its Reversal in Aging and Neurodegeneration Involve miR-183/96/182 Biogenesis. Mol. Neurobiol. 2019, 56, 3451–3462. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Li, L.; Zhang, M.-D.; Gonzales, C.B.; Parisien, M.; Belfer, I.; Usoskin, D.; Abdo, H.; Furlan, A.; Häring, M.; et al. miR-183 cluster scales mechanical pain sensitivity by regulating basal and neuropathic pain genes. Science 2017, 356, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.A.; Quint, E.; Glazier, A.M.; Fuchs, H.; De Angelis, M.H.; Langford, C.; Van Dongen, S.; Abreu-Goodger, C.; Piipari, M.; Redshaw, N.; et al. An ENU-induced mutation of miR-96 associated with progressive hearing loss in mice. Nat. Genet. 2009, 41, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Mencía, A.; Modamio-Høybjør, S.; Redshaw, N.; Morín, M.; Mayo-Merino, F.; Olavarrieta, L.; Aguirre, L.A.; Del Cadtillo, I.; Steel, K.P.; Dalmay, T.; et al. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat. Genet. 2009, 41, 609–613. [Google Scholar] [CrossRef]
- Dambal, S.; Shah, M.; Mihelich, B.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef]
- Jin, Z.-B.; Hirokawa, G.; Gui, L.; Takahashi, R.; Osakada, F.; Hiura, Y.; Takahashi, M.; Yasuhara, O.; Iwai, N. Targeted deletion of miR-182, an abundant retinal microRNA. Mol. Vis. 2009, 15, 523–533. [Google Scholar]
- Fogerty, J.; Stepanyan, R.; Cianciolo, L.T.; Tooke, B.P.; Perkins, B.D. Genomic non-redundancy of the mir-183/96/182 cluster and its requirement for hair cell maintenance. Sci. Rep. 2019, 9, 10302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Ma, L.; Xi, K.; Zhang, W.; Fan, D. MicroRNA-183 Suppresses Neuropathic Pain and Expression of AMPA Receptors by Targeting mTOR/VEGF Signaling Pathway. Cell. Physiol. Biochem. 2017, 41, 181–192. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiltschewskij, D.J.; Geaghan, M.P.; Cairns, M.J. Characterising the Transcriptional and Translational Impact of the Schizophrenia-Associated miR-1271-5p in Neuronal Cells. Cells 2020, 9, 1014. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041014
Kiltschewskij DJ, Geaghan MP, Cairns MJ. Characterising the Transcriptional and Translational Impact of the Schizophrenia-Associated miR-1271-5p in Neuronal Cells. Cells. 2020; 9(4):1014. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041014
Chicago/Turabian StyleKiltschewskij, Dylan J., Michael P. Geaghan, and Murray J. Cairns. 2020. "Characterising the Transcriptional and Translational Impact of the Schizophrenia-Associated miR-1271-5p in Neuronal Cells" Cells 9, no. 4: 1014. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041014