Ca2+–Calmodulin Dependent Wound Repair in Dictyostelium Cell Membrane


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Plasmids and Transformation
2.3. Chamber Preparation
2.4. Wounding and Microscopy
2.5. Measurement of the Influx of Propidium Iodide, FM1-43, and Ca2+
2.6. Ca–EGTA Buffer
2.7. Inhibitors
2.8. Statistical Analysis
3. Results
3.1. Influx of Ca2+ through the Wound Pore is Essential for Wound Repair
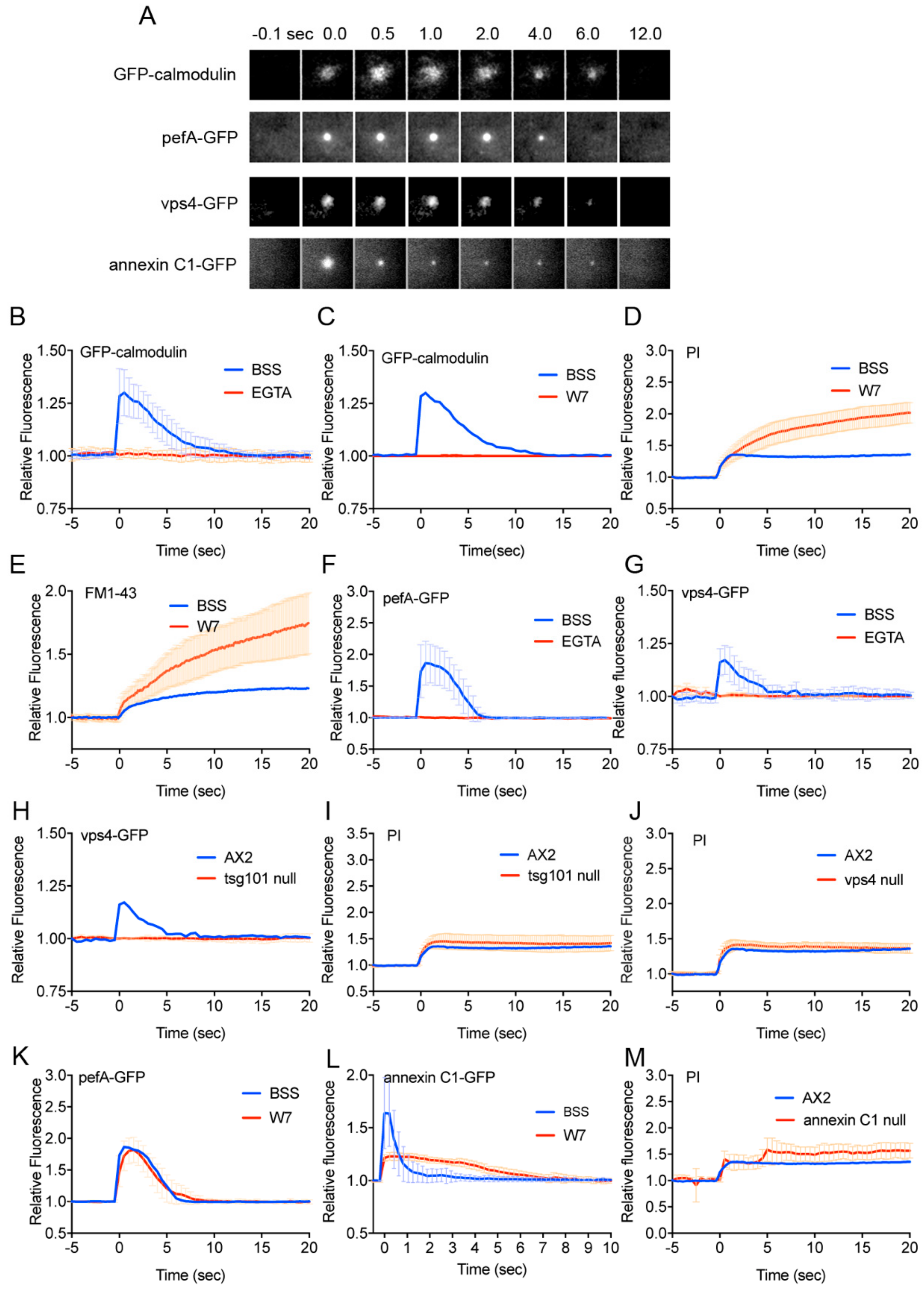
3.2. Calmodulin, ESCRT, and Annexin Quickly Accumulate at the Wound Site
3.3. Actin Accumulation at Wound Site is Essential for Wound Repair
3.4. Membrane Plug is Formed at the Wound Site
3.5. Membrane Vesicles for the Plug are De Novo Generated at the Wound Site
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sonnemann, K.J.; Bement, W.M. Wound repair: Toward understanding and integration of single–cell and multicellular wound responses. Annu. Rev. Cell Dev. Biol. 2011, 27, 237–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Dominguez, A.N.M.; Decker, J.R.; Hull, A.J.; Verboon, J.M.; Parkhurst, S.M. Into the breach: How cells cope with wounds. Open Biol. 2018, 8, 180135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, A.C.; McNeil, A.K.; Xiong, F.; Xiong, W.C.; McNeil, P.L. A novel cellular defect in diabetes: Membrane repair failure. Diabetes 2011, 60, 3034–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin–deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.B.; Lemckert, F.A.; Zheng, X.F.; Tran, J.; Evesson, F.J.; Hawkes, J.M.; Lek, A.; Street, N.E.; Lin, P.; Clarke, N.F.; et al. Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T–system with stretch. J. Neuropathol. Exp. Neurol. 2011, 70, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53–mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra36. [Google Scholar] [CrossRef] [Green Version]
- Labazi, M.; McNeil, A.K.; Kurtz, T.; Lee, T.C.; Pegg, R.B.; Angeli, J.P.F.; Conrad, M.; McNeil, P.L. The antioxidant requirement for plasma membrane repair in skeletal muscle. Free. Radic. Biol. Med. 2015, 84, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Razzell, W.; Wood, W.; Martin, P. Swatting flies: Modelling wound healing and inflammation in Drosophila. Dis. Model Mech. 2011, 4, 569–574. [Google Scholar] [CrossRef] [Green Version]
- Abreu–Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J. Cell. Biol. 2011, 193, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Abreu–Blanco, M.T.; Verboon, J.M.; Liu, R.; Watts, J.J.; Parkhurst, S.M. Drosophila embryos close epithelial wounds using a combination of cellular protrusions and an actomyosin purse string. J. Cell Sci. 2012, 125, 5984–5997. [Google Scholar] [CrossRef] [Green Version]
- Abreu–Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Coordination of Rho family GTPase activities to orchestrate cytoskeleton responses during cell wound repair. Curr. Biol. 2014, 24, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, K.; Saeki, Y.; Yoshida, S.; Tanaka, K.; Pellman, D. Proteasomal degradation resolves competition between cell polarization and cellular wound healing. Cell 2012, 150, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Chisholm, A.D. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell 2014, 31, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, S.; Soto, X.; Woolner, S.; Amaya, E. ERK and phosphoinositide 3-kinase temporally coordinate different modes of actin–based motility during embryonic wound healing. J. Cell Sci. 2013, 126, 5005–5017. [Google Scholar] [CrossRef] [Green Version]
- Bouter, A.; Gounou, C.; Berat, R.; Tan, S.; Gallois, B.; Granier, T.; d’Estaintot, B.L.; Poschl, E.; Brachvogel, B.; Brisson, A.R. Annexin–A5 assembled into two–dimensional arrays promotes cell membrane repair. Nat. Commun. 2011, 2, 270. [Google Scholar] [CrossRef] [Green Version]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sonder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [Green Version]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.Y.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin–dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef]
- Horn, A.; Van der Meulen, J.H.; Defour, A.; Hogarth, M.; Sreetama, S.C.; Reed, A.; Scheffer, L.; Chandel, N.S.; Jaiswal, J.K. Mitochondrial redox signaling enables repair of injured skeletal muscle cells. Sci. Signal 2017, 10, eaaj1978. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.; Idone, V.; Devlin, C.; Fernandes, M.C.; Flannery, A.; He, X.; Schuchman, E.; Tabas, I.; Andrews, N.W. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 2010, 189, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Pervin, M.S.; Itoh, G.; Talukder, M.S.U.; Fujimoto, K.; Morimoto, Y.V.; Tanaka, M.; Ueda, M.; Yumura, S. A study of wound repair in Dictyostelium cells by using novel laserporation. Sci. Rep. 2018, 8, 7969. [Google Scholar] [CrossRef]
- Bi, G.Q.; Alderton, J.M.; Steinhardt, R.A. Calcium–regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 1995, 131, 1747–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Vogel, S.S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [PubMed]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large plasma membrane disruptions are rapidly resealed by Ca2+–dependent vesicle–vesicle fusion events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Davenport, N.R.; Sonnemann, K.J.; Eliceiri, K.W.; Bement, W.M. Membrane dynamics during cellular wound repair. Mol. Biol. Cell 2016, 27, 2272–2285. [Google Scholar] [CrossRef]
- Reddy, A.; Caler, E.V.; Andrews, N.W. Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 2001, 106, 157–169. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef]
- Moe, A.M.; Golding, A.E.; Bement, W.M. Cell healing: Calcium, repair and regeneration. Semin. Cell Dev. Biol. 2015, 45, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef] [Green Version]
- Roostalu, U.; Strahle, U. In vivo imaging of molecular interactions at damaged sarcolemma. Dev. Cell 2012, 22, 515–529. [Google Scholar] [CrossRef] [Green Version]
- Swaggart, K.A.; Demonbreun, A.R.; Vo, A.H.; Swanson, K.E.; Kim, E.Y.; Fahrenbach, J.P.; Holley–Cuthrell, J.; Eskin, A.; Chen, Z.; Squire, K.; et al. Annexin A6 modifies muscular dystrophy by mediating sarcolemmal repair. Proc. Natl. Acad. Sci. USA 2014, 111, 6004–6009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jaattela, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [Green Version]
- McNeil, A.K.; Rescher, U.; Gerke, V.; McNeil, P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 2006, 281, 35202–35207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez–Jimenez, A.T.; Cardenal–Munoz, E.; Leuba, F.; Gerstenmaier, L.; Barisch, C.; Hagedorn, M.; King, J.S.; Soldati, T. The ESCRT and autophagy machineries cooperate to repair ESX–1–dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PLoS Pathog. 2018, 14, e1007501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radulovic, M.; Schink, K.O.; Wenzel, E.M.; Nahse, V.; Bongiovanni, A.; Lafont, F.; Stenmark, H. ESCRT–mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018, 37, e99753. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT–dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.T.; McNeil, P.L. Membrane repair: Mechanisms and pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Miyake, K.; McNeil, P.L.; Suzuki, K.; Tsunoda, R.; Sugai, N. An actin barrier to resealing. J. Cell Sci. 2001, 114, 3487–3494. [Google Scholar]
- Bement, W.M.; Mandato, C.A.; Kirsch, M.N. Wound–induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr. Biol. 1999, 9, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Steinhardt, R.A. Nonmuscle myosin IIA and IIB have distinct functions in the exocytosis–dependent process of cell membrane repair. Mol. Biol. Cell 2004, 15, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S.; Hashima, S.; Muranaka, S. Myosin II does not contribute to wound repair in Dictyostelium cells. Biol. Open 2014, 3, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henson, J.H.; Nazarian, R.; Schulberg, K.L.; Trabosh, V.A.; Kolnik, S.E.; Burns, A.R.; McPartland, K.J. Wound closure in the lamellipodia of single cells: Mediation by actin polymerization in the absence of an actomyosin purse string. Mol. Biol. Cell 2002, 13, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumura, S. Myosin II dynamics and cortical flow during contractile ring formation in Dictyostelium cells. J. Cell Biol. 2001, 154, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Pramanik, M.K.; Yumura, S. Microtubule-Mediated Inositol Lipid Signaling Plays Critical Roles in Regulation of Blebbing. PLoS ONE 2015, 10, e0137032. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kikuchi, T.; Uno, H.; Okita, K.; Kitanishi-Yumura, T.; Yumura, S. Turnover and flow of the cell membrane for cell migration. Sci. Rep. 2017, 7, 12970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, K.S.; Kitanishi-Yumura, T.; Yumura, S. Myosin II contributes to the posterior contraction and the anterior extension during the retraction phase in migrating Dictyostelium cells. J. Cell Sci. 2003, 116, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S. A novel low-power laser–mediated transfer of foreign molecules into cells. Sci. Rep. 2016, 6, 22055. [Google Scholar] [CrossRef]
- Yumura, S.; Matsuzaki, R.; Kitanishi–Yumura, T. Introduction of macromolecules into living Dictyostelium cells by electroporation. Cell Struct. Funct. 1995, 20, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Yumura, S.; Itoh, G.; Kikuta, Y.; Kikuchi, T.; Kitanishi-Yumura, T.; Tsujioka, M. Cell–scale dynamic recycling and cortical flow of the actin–myosin cytoskeleton for rapid cell migration. Biol. Open 2013, 2, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Pervin, M.S.; Yumura, S. Manipulation of cell migration by laserporation-induced local wounding. Sci. Rep. 2019, 9, 4291. [Google Scholar] [CrossRef]
- Yumura, S.; Mori, H.; Fukui, Y. Localization of actin and myosin for the study of ameboid movement in Dictyostelium using improved immunofluorescence. J. Cell Biol. 1984, 99, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, L.L.; Sreetama, S.C.; Sharma, N.; Medikayala, S.; Brown, K.J.; Defour, A.; Jaiswal, J.K. Mechanism of Ca2+–triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun. 2014, 5, 5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellgren, R.L. A plasma membrane wound proteome: Reversible externalization of intracellular proteins following reparable mechanical damage. J. Biol. Chem. 2010, 285, 36597–36607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, N.; Schwartz, J.M.; Kohler, J.; Becker, M.; Schwarz, H.; Gerisch, G. Golvesin-GFP fusions as distinct markers for Golgi and post–Golgi vesicles in Dictyostelium cells. Biol. Cell 2000, 92, 495–511. [Google Scholar] [CrossRef]
- Karakesisoglou, I.; Janssen, K.P.; Eichinger, L.; Noegel, A.A.; Schleicher, M. Identification of a suppressor of the Dictyostelium profilin–minus phenotype as a CD36/LIMP–II homologue. J. Cell Biol. 1999, 145, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Sattler, N.; Bosmani, C.; Barisch, C.; Gueho, A.; Gopaldass, N.; Dias, M.; Leuba, F.; Bruckert, F.; Cosson, P.; Soldati, T. Functions of the Dictyostelium LIMP–2 and CD36 homologues in bacteria uptake, phagolysosome biogenesis and host cell defence. J. Cell Sci. 2018, 131, jcs218040. [Google Scholar] [CrossRef] [Green Version]
- Muller-Taubenberger, A.; Lupas, A.N.; Li, H.; Ecke, M.; Simmeth, E.; Gerisch, G. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 2001, 20, 6772–6782. [Google Scholar] [CrossRef] [Green Version]
- Mandato, C.A.; Bement, W.M. Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Curr. Biol. 2003, 13, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Togo, T. Disruption of the plasma membrane stimulates rearrangement of microtubules and lipid traffic toward the wound site. J. Cell Sci. 2006, 119, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Carmeille, R.; Bouvet, F.; Tan, S.; Croissant, C.; Gounou, C.; Mamchaoui, K.; Mouly, V.; Brisson, A.R.; Bouter, A. Membrane repair of human skeletal muscle cells requires Annexin-A5. Biochim. Biophys. Acta 2016, 1863, 2267–2279. [Google Scholar] [CrossRef]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT machinery is required for plasma membrane repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, S.P.; Boye, T.L.; Nylandsted, J. Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin. Cell Dev. Biol. 2015, 45, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Goddard, R.H.; La, C.J.W. Calmodulin and wound healing in the coenocytic green alga Ernodesmis verticillata (Kutzing) Borgesen: Immunofluorescence and effects of antagonists. Planta 1991, 183, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.Q.; Morris, R.L.; Liao, G.; Alderton, J.M.; Scholey, J.M.; Steinhardt, R.A. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J. Cell Biol. 1997, 138, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, N.R.; Bement, W.M. Cell repair: Revisiting the patch hypothesis. Commun. Integr. Biol. 2016, 9, e1253643. [Google Scholar] [CrossRef]
- Miyanaga, Y.; Matsuoka, S.; Ueda, M. Single–molecule imaging techniques to visualize chemotactic signaling events on the membrane of living Dictyostelium cells. Methods Mol. Biol. 2009, 571, 417–435. [Google Scholar]
- Shibutani, S.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Mandato, C.A.; Bement, W.M. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J. Cell Biol. 2001, 154, 785–797. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H.J. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound–healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [Green Version]
- Corrotte, M.; Almeida, P.E.; Tam, C.; Castro-Gomes, T.; Fernandes, M.C.; Millis, B.A.; Cortez, M.; Miller, H.; Song, W.; Maugel, T.K.; et al. Caveolae internalization repairs wounded cells and muscle fibers. eLife 2013, 2, e00926. [Google Scholar] [CrossRef]
- Miyake, K.; McNeil, P.L. Vesicle accumulation and exocytosis at sites of plasma membrane disruption. J. Cell Biol. 1995, 131, 1737–1745. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talukder, M.S.U.; Pervin, M.S.; Tanvir, M.I.O.; Fujimoto, K.; Tanaka, M.; Itoh, G.; Yumura, S. Ca2+–Calmodulin Dependent Wound Repair in Dictyostelium Cell Membrane. Cells 2020, 9, 1058. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041058
Talukder MSU, Pervin MS, Tanvir MIO, Fujimoto K, Tanaka M, Itoh G, Yumura S. Ca2+–Calmodulin Dependent Wound Repair in Dictyostelium Cell Membrane. Cells. 2020; 9(4):1058. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041058
Chicago/Turabian StyleTalukder, Md. Shahabe Uddin, Mst. Shaela Pervin, Md. Istiaq Obaidi Tanvir, Koushiro Fujimoto, Masahito Tanaka, Go Itoh, and Shigehiko Yumura. 2020. "Ca2+–Calmodulin Dependent Wound Repair in Dictyostelium Cell Membrane" Cells 9, no. 4: 1058. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9041058