Vγ9Vδ2 T Cells: Can We Re-Purpose a Potent Anti-Infection Mechanism for Cancer Therapy?

 and
and
Abstract
:1. From Coley’s Toxin to Pattern Recognition Receptors
2. BTN3A1 is a PAMP Receptor
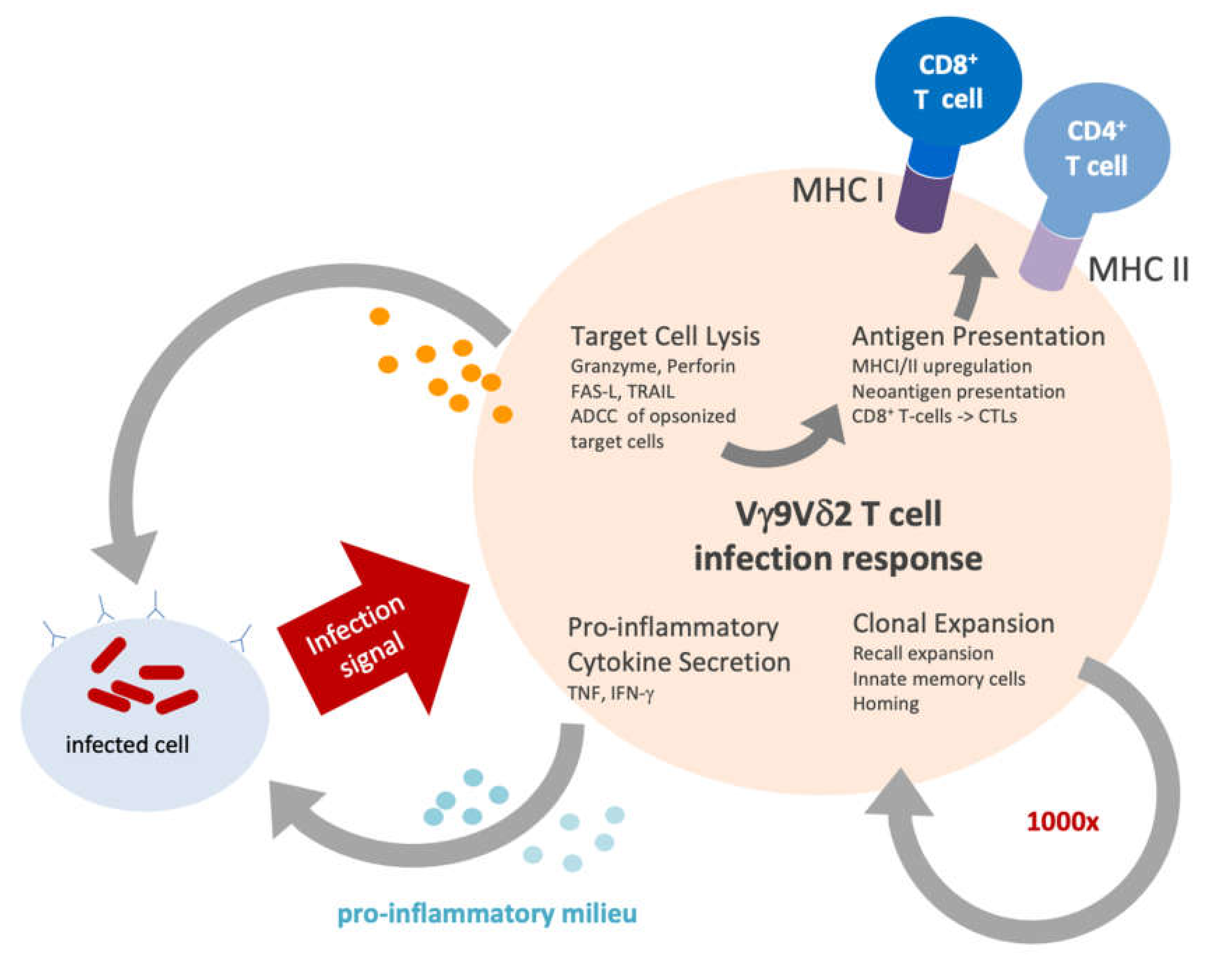
3. Vγ9Vδ2 T Cells in Infection
4. Vγ9Vδ2T Cells in Cancer
5. Plasticity of Vγ9Vδ2 T Cells
5.1. Cytotoxicity
5.2. Antigen Presentation
5.3. Clonal Expansion
5.4. Suppressor Functionality of γδ T Cells
6. Targeting Vγ9Vδ2 T Cells in Cancer Therapy
6.1. Adoptive T Cell-Transfer and In Vivo Stimulation
6.2. Chimeric Antigen Receptor T Cells (CAR-Ts)
6.3. BTN3A Agonistic Antibody
6.4. T Cell Engager Approaches: γδ T Cell Engagers (γδ TcE)
7. Vγ9Vδ2 T-Cell Functionalities in Tumor Targeting
8. Towards Precision Immune-Oncology
9. Preclinical Modeling

{kind=link}
{kind=link}
| Reference | Number of Transferred Cells | Route | Cell Source | Administration | In Vivo BTN3A Activation | Exogenous Cytokine Administration | Tumor | Mouse Strains |
|---|---|---|---|---|---|---|---|---|
| [122] | 3 × 107 Vγ9Vδ2 | i.v. | PBMC | single | 20.1 mAb | IL-15/IL-15ra (RLI) | Primary AML, U937 | NSG |
| [161] | 1 × 106 γδ T cells | i.p. | γδTILs/TALs | single | none | IL-2 | Daudi/SKOV3 | BALB/c nude |
| [162] | 2 x107 Vγ9Vδ2 | i.p. | PBMC | repetitive | zoledronate | IL-2 | MM1 CML | SCID |
| [163] | 2 x107 γδ T cells | i.v. | PBMC | repetitive | none | none | 2LMP | SCID |
| [153] | 5 x106 Vγ9Vδ2 | i.v. | PBMC | repetitive | zoledronate | no | SH-SY-5Y | BALB/c nude |
| [164] | 5 × 106 γδ T cells | s.c. | PBMC | single | none | no | NCI-H460 | SCID |
| [154] | 4 x107 Vγ9Vδ2 | i.v. | PBMC | single | alendronate | no | A375 | SCID |
| [156] | 4 x107 Vγ9Vδ2 | i.v. | PBMC | single | no | no | U937 | NOG |
| [149] | 1 x107 Vγ9Vδ2 | intracranial | PBMC | single and repetitive | zoledronate | none | U-87MG/orthotopic GBM | NSG |
| [147] | various | i.p. | PBMC | repetitive | alendronate | IL-2 | MeWo PancTu1 | SCID |
| [165] | 2 × 106 Vγ9Vδ2 | i.v. | PBMC | repetitive | none | IL-2 | Autolog. melanoma | CB.17 SCID |
| [146] | 1 x107 Vγ9Vδ2 | i.v. | PBMC | single | aledronate zoledronate | no | MDA-MB-231-hNIS.GFP | NSG |
| [157] | 1x 107 Vγ9Vδ2 enriched PBMCs | i.p. | PBMC | repetitive | none | no | Daudi | SCID |
| [150] | 1 × 106 Vγ9Vδ2 | i.v. | PBMC | single and repetitive | pamidronate | no | OVCAR-3 | NSG |
| [134] | 1.5 -3 × 105 Vγ9Vδ2 | s.c. | PBMC | repetitive | Zoledronate [(Her2)2xVγ9] | IL-2 | PancTu-I (PDAC) | SCIDbeige |
| [155] | 1 x107 Vγ9Vδ2 | i.p. | PBMC | single and repetitive | aledronate zoledronate | no | SKOV-3 IGROV | SCID |
| [151] | 1 × 106 Vγ9Vδ2 | i.v. | PBMC | single and repetitive | pamidronate | no | PC3 | NSG |
| [158] | 1 × 107 PBMC +/- Vγ9Vδ2 | i.v. | PBMC | repetitive | pamidronate | no | EBV induced B cell lymphoma | Rag2-/- γc -/- |
| [50] | 1 x107 Vγ9Vδ2 | intravesicular | PBMC | single | zoledronate | none | UM-UC-3 | SCID |
| [152] | 5 × 107 pan γδ T cells | i.v. | PBMC | single and repetitive | none | no | CNE2 | BALB/c nude |
| [159] | 1 x107 Vγ9Vδ2 | i.v. | PBMC | single | no | no | EBV induced B cell lymphoma | NSG |
10. Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Decker, W.K.; Safdar, A. Bioimmunoadjuvants for the treatment of neoplastic and infectious disease: Coley’s legacy revisited. Cytokine Growth Factor Rev. 2009, 20, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharm. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Lamm, D.L.; Blumenstein, B.A.; Crawford, E.D.; Montie, J.E.; Scardino, P.; Grossman, H.B.; Stanisic, T.H.; Smith, J.A.; Sullivan, J.; Sarosdy, M.F. A randomized trial of intravesical doxorubicin and immunotherapy with bacille Calmette-Guérin for transitional-cell carcinoma of the bladder. N. Engl. J. Med. 1991, 325, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate immune pattern recognition: A cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.J.; Ahn, B.Y.; Lee, N.G.; Lee, D.H.; Kim, D.-S. A combination of E. coli DNA fragments and modified lipopolysaccharides as a cancer immunotherapy. Vaccine 2006, 24, 5862–5871. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef]
- Wei, M.Q.; Mengesha, A.; Good, D.; Anné, J. Bacterial targeted tumour therapy-dawn of a new era. Cancer Lett. 2008, 259, 16–27. [Google Scholar] [CrossRef]
- Braunstein, M.J.; Kucharczyk, J.; Adams, S. Targeting Toll-Like Receptors for Cancer Therapy. Target Oncol 2018, 13, 583–598. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Parker, A.E.; O’Neill, L.A.J. Targeting Toll-like receptors: Emerging therapeutics? Nat. Rev. Drug Discov. 2010, 9, 293–307. [Google Scholar] [CrossRef]
- Mullard, A. Can innate immune system targets turn up the heat on “cold” tumours? Nat. Rev. Drug Discov. 2018, 17, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.A.; Chen, H.-C.; Price, A.J.; Keeble, A.H.; Davey, M.S.; James, L.C.; Eberl, M.; Trowsdale, J. Activation of human γδ T cells by cytosolic interactions of BTN3A1 with soluble phosphoantigens and the cytoskeletal adaptor periplakin. J. Immunol. 2015, 194, 2390–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harly, C.; Guillaume, Y.; Nedellec, S.; Peigne, C.M.; Monkkonen, H.; Monkkonen, J.; Li, J.; Kuball, J.; Adams, E.J.; Netzer, S.; et al. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human T-cell subset. Blood 2012, 120, 2269–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandstrom, A.; Peigné, C.-M.; Léger, A.; Crooks, J.E.; Konczak, F.; Gesnel, M.-C.; Breathnach, R.; Bonneville, M.; Scotet, E.; Adams, E.J. The Intracellular B30.2 Domain of Butyrophilin 3A1 Binds Phosphoantigens to Mediate Activation of Human Vγ9Vδ2 T Cells. Immunity 2014, 40, 490–500. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Gu, S.; Nawrocka, W.; Adams, E.J. Sensing of Pyrophosphate Metabolites by Vγ9Vδ2 T Cells. Front. Immunol. 2014, 5, 688. [Google Scholar]
- Vavassori, S.; Kumar, A.; Wan, G.S.; Ramanjaneyulu, G.S.; Cavallari, M.; El Daker, S.; Beddoe, T.; Theodossis, A.; Williams, N.K.; Gostick, E.; et al. Butyrophilin 3A1 binds phosphorylated antigens and stimulates human γδ T cells. Nat. Immunol. 2013, 14, 908–916. [Google Scholar] [CrossRef]
- Gu, S.; Borowska, M.T.; Boughter, C.T.; Adams, E.J. Butyrophilin3A proteins and Vγ9Vδ2 T cell activation. Semin. Cell Dev. Biol. 2018, 84, 65–74. [Google Scholar] [CrossRef]
- Peigné, C.-M.; Léger, A.; Gesnel, M.-C.; Konczak, F.; Olive, D.; Bonneville, M.; Breathnach, R.; Scotet, E. The Juxtamembrane Domain of Butyrophilin BTN3A1 Controls Phosphoantigen-Mediated Activation of Human Vγ9Vδ2 T Cells. J. Immunol. 2017, 198, 4228–4234. [Google Scholar] [CrossRef] [Green Version]
- Sebestyen, Z.; Scheper, W.; Vyborova, A.; Gu, S.; Rychnavska, Z.; Schiffler, M.; Cleven, A.; Chéneau, C.; van Noorden, M.; Peigné, C.-M.; et al. RhoB Mediates Phosphoantigen Recognition by Vγ9Vδ2 T Cell Receptor. CellReports 2016, 15, 1973–1985. [Google Scholar] [CrossRef] [Green Version]
- De Libero, G.; Lau, S.-Y.; Mori, L. Phosphoantigen Presentation to TCR γδ Cells, a Conundrum Getting Less Gray Zones. Front. Immunol. 2014, 5, 679. [Google Scholar]
- Gruenbacher, G.; Gander, H.; Rahm, A.; Idzko, M.; Nussbaumer, O.; Thurnher, M. Ecto-ATPase CD39 Inactivates Isoprenoid-Derived Vγ9Vδ2 T Cell Phosphoantigens. CellReports 2016, 16, 444–456. [Google Scholar] [CrossRef] [Green Version]
- Riaño, F.; Karunakaran, M.M.; Starick, L.; Li, J.; Scholz, C.J.; Kunzmann, V.; Olive, D.; Amslinger, S.; Herrmann, T. Vγ9Vδ2 TCR-activation by phosphorylated antigens requires butyrophilin 3 A1 (BTN3A1) and additional genes on human chromosome 6. Eur. J. Immunol. 2014, 44, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science 2020, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, M.M.; Willcox, C.R.; Salim, M.; Paletta, D.; Fichtner, A.S.; Noll, A.; Starick, L.; Nöhren, A.; Begley, C.R.; Berwick, K.A.; et al. Butyrophilin-2A1 Directly Binds Germline-Encoded Regions of the Vγ9Vδ2 TCR and Is Essential for Phosphoantigen Sensing. Immunity 2020, 52, 487–498.e6. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.E.; Willcox, C.R. γδ TCR ligands: The quest to solve a 500-million-year-old mystery. Nat. Immunol. 2019, 20, 1–8. [Google Scholar] [CrossRef]
- Melandri, D.; Zlatareva, I.; Chaleil, R.A.G.; Dart, R.J.; Chancellor, A.; Nussbaumer, O.; Polyakova, O.; Roberts, N.A.; Wesch, D.; Kabelitz, D.; et al. The γδTCR combines innate immunity with adaptive immunity by utilizing spatially distinct regions for agonist selection and antigen responsiveness. Nat. Immunol. 2018, 19, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.M.R. Gammadelta T cells: Innately adaptive immune cells? Int. Rev. Immunol. 2013, 32, 223–248. [Google Scholar] [CrossRef]
- Perera, M.K.; Carter, R.; Goonewardene, R.; Mendis, K.N. Transient increase in circulating gamma/delta T cells during Plasmodium vivax malarial paroxysms. J. Exp. Med. 1994, 179, 311–315. [Google Scholar] [CrossRef] [Green Version]
- Raziuddin, S.; Telmasani, A.W.; el-Hag el-Awad, M.; al-Amari, O.; al-Janadi, M. Gamma delta T cells and the immune response in visceral leishmaniasis. Eur. J. Immunol. 1992, 22, 1143–1148. [Google Scholar] [CrossRef]
- Jouen-Beades, F.; Paris, E.; Dieulois, C.; Lemeland, J.F.; Barre-Dezelus, V.; Marret, S.; Humbert, G.; Leroy, J.; Tron, F. In vivo and in vitro activation and expansion of gammadelta T cells during Listeria monocytogenes infection in humans. Infect. Immun. 1997, 65, 4267–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, T.; Maeda, T.; Takahashi, H.; Yonaha, F.; Sakamoto, A.; Tomioka, H.; Koike, T.; YOSHIDA, S. Predominant expansion of V gamma 9/V delta 2 T cells in a tularemia patient. Infect. Immun. 1992, 60, 2554–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, O.; Guirado, E.; Gordillo, S.; Díaz, J.; Tapia, G.; Vilaplana, C.; Ariza, A.; Ausina, V.; Cardona, P.-J. Intragranulomatous necrosis in lungs of mice infected by aerosol with Mycobacterium tuberculosis is related to bacterial load rather than to any one cytokine or T cell type. Microbes Infect. 2006, 8, 628–636. [Google Scholar] [CrossRef]
- Behar, S.M.; Porcelli, S.A. CD1-restricted T cells in host defense to infectious diseases. Curr. Top. Microbiol. Immunol. 2007, 314, 215–250. [Google Scholar] [PubMed]
- Kabelitz, D.; Bender, A.; Prospero, T.; Wesselborg, S.; Janssen, O.; Pechhold, K. The primary response of human gamma/delta + T cells to Mycobacterium tuberculosis is restricted to V gamma 9-bearing cells. J. Exp. Med. 1991, 173, 1331–1338. [Google Scholar]
- Havlir, D.V.; Ellner, J.J.; Chervenak, K.A.; Boom, W.H. Selective expansion of human gamma delta T cells by monocytes infected with live Mycobacterium tuberculosis. J. Clin. Invest. 1991, 87, 729–733. [Google Scholar] [CrossRef]
- Ryan-Payseur, B.; Frencher, J.; Shen, L.; Chen, C.Y.; Huang, D.; Chen, Z.W. Multieffector-Functional Immune Responses of HMBPP-Specific Vγ2Vδ2 T Cells in Nonhuman Primates Inoculated with Listeria monocytogenes ΔactA prfA*. J. Immunol. 2012, 189, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Zhou, D.; Qiu, L.; Lai, X.; Simon, M.; Shen, L.; Kou, Z.; Wang, Q.; Jiang, L.; Estep, J.; et al. Adaptive immune response of Vgamma2Vdelta2+ T cells during mycobacterial infections. Science 2002, 295, 2255–2258. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 1–12. [Google Scholar] [CrossRef]
- Tosolini, M.; Pont, F.; Poupot, M.; Vergez, F.; Nicolau-Travers, M.-L.; Vermijlen, D.; Sarry, J.E.; Dieli, F.; Fournié, J.-J. Assessment of tumor-infiltrating TCRVγ9Vδ2 γδ lymphocyte abundance by deconvolution of human cancers microarrays. Oncoimmunology 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Scotet, E.; Martinez, L.O.; Grant, E.; Barbaras, R.; Jenö, P.; Guiraud, M.; Monsarrat, B.; Saulquin, X.; Maillet, S.; Estève, J.-P.; et al. Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity 2005, 22, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Chen, H.; Mo, C.; Cui, L.; He, W. Ectopically expressed human tumor biomarker MutS homologue 2 is a novel endogenous ligand that is recognized by human γδ T cells to induce innate anti-tumor/virus immunity. J. Biol. Chem. 2012, 287, 16812–16819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T Cell Receptor γδ Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Morita, C.T.; Jin, C.; Sarikonda, G.; Wang, H. Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vgamma2Vdelta2 T cells: Discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol. Rev. 2007, 215, 59–76. [Google Scholar] [PubMed]
- Benzaïd, I.; Mönkkönen, H.; Stresing, V.; Bonnelye, E.; Green, J.; Mönkkönen, J.; Touraine, J.-L.; Clézardin, P. High phosphoantigen levels in bisphosphonate-treated human breast tumors promote Vgamma9Vdelta2 T-cell chemotaxis and cytotoxicity in vivo. Cancer Res. 2011, 71, 4562–4572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauhiainen, M.; Mönkkönen, H.; Räikkönen, J.; Mönkkönen, J.; Auriola, S. Analysis of endogenous ATP analogs and mevalonate pathway metabolites in cancer cell cultures using liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2967–2975. [Google Scholar] [CrossRef]
- Bryant, N.L.; Suarez-Cuervo, C.; Gillespie, G.Y.; Markert, J.M.; Nabors, L.B.; Meleth, S.; Lopez, R.D.; Lamb, L.S. Characterization and immunotherapeutic potential of gammadelta T-cells in patients with glioblastoma. Neuro-Oncol. 2009, 11, 357–367. [Google Scholar] [CrossRef]
- Rey, J.; Veuillen, C.; Vey, N.; Bouabdallah, R.; Olive, D. Natural killer and γδ T cells in haematological malignancies: Enhancing the immune effectors. Trends Mol. Med. 2009, 15, 275–284. [Google Scholar] [CrossRef]
- Todaro, M.; D’Asaro, M.; Caccamo, N.; Iovino, F.; Francipane, M.G.; Meraviglia, S.; Orlando, V.; La Mendola, C.; Gulotta, G.; Salerno, A.; et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J. Immunol. 2009, 182, 7287–7296. [Google Scholar] [CrossRef] [Green Version]
- Yuasa, T.; Sato, K.; Ashihara, E.; Takeuchi, M.; Maita, S.; Tsuchiya, N.; Habuchi, T.; Maekawa, T.; Kimura, S. Intravesical administration of gammadelta T cells successfully prevents the growth of bladder cancer in the murine model. Cancer Immunol. Immunother. 2009, 58, 493–502. [Google Scholar] [CrossRef]
- Banerjee, S.; Tian, T.; Wei, Z.; Shih, N.; Feldman, M.D.; Peck, K.N.; DeMichele, A.M.; Alwine, J.C.; Robertson, E.S. Distinct Microbial Signatures Associated with Different Breast Cancer Types. Front. Microbiol. 2018, 9, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogdill, A.P.; Gaudreau, P.O.; Arora, R.; Gopalakrishnan, V.; Wargo, J.A. The Impact of Intratumoral and Gastrointestinal Microbiota on Systemic Cancer Therapy. Trends Immunol. 2018, 39, 900–920. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Tian, T.; Wei, Z.; Peck, K.N.; Shih, N.; Chalian, A.A.; O’Malley, B.W.; Weinstein, G.S.; Feldman, M.D.; Alwine, J.; et al. Microbial Signatures Associated with Oropharyngeal and Oral Squamous Cell Carcinomas. Sci. Rep. 2017, 7, 135–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redelman-Sidi, G.; Iyer, G.; Solit, D.B.; Glickman, M.S. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res. 2013, 73, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef] [Green Version]
- Healy, A.R.; Nikolayevskiy, H.; Patel, J.R.; Crawford, J.M.; Herzon, S.B. A Mechanistic Model for Colibactin-Induced Genotoxicity. J. Am. Chem. Soc. 2016, 138, 15563–15570. [Google Scholar] [CrossRef] [Green Version]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host. Microbe. 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Spada, F.M.; Grant, E.P.; Peters, P.J.; Sugita, M.; Melián, A.; Leslie, D.S.; Lee, H.K.; van Donselaar, E.; Hanson, D.A.; Krensky, A.M.; et al. Self-recognition of CD1 by gamma/delta T cells: Implications for innate immunity. J. Exp. Med. 2000, 191, 937–948. [Google Scholar] [CrossRef]
- Caccamo, N.; Dieli, F.; Meraviglia, S.; Guggino, G.; Salerno, A. Gammadelta T cell modulation in anticancer treatment. Curr. Cancer Drug Targets 2010, 10, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Dieli, F.; Troye-Blomberg, M.; Ivanyi, J.; Fournie, J.J.; Krensky, A.M.; Bonneville, M.; Peyrat, M.A.; Caccamo, N.; Sireci, G.; Salerno, A. Granulysin-dependent killing of intracellular and extracellular Mycobacterium tuberculosis by Vgamma9/Vdelta2 T lymphocytes. J. Infect. Dis. 2001, 184, 1082–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, C.T.; Abate, G.; Sakala, I.G.; Xia, M.; Truscott, S.M.; Eickhoff, C.S.; Linn, R.; Blazevic, A.; Metkar, S.S.; Peng, G.; et al. Granzyme A produced by γ(9)δ(2) T cells induces human macrophages to inhibit growth of an intracellular pathogen. Plos Pathog. 2013, 9, e1003119. [Google Scholar] [CrossRef] [PubMed]
- Oberg, H.-H.; Janitschke, L.; Sulaj, V.; Weimer, J.; Gonnermann, D.; Hedemann, N.; Arnold, N.; Kabelitz, D.; Peipp, M.; Bauerschlag, D.; et al. Bispecific antibodies enhance tumor-infiltrating T cell cytotoxicity against autologous HER-2-expressing high-grade ovarian tumors. J. Leukoc. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Couzi, L.; Pitard, V.; Sicard, X.; Garrigue, I.; Hawchar, O.; Merville, P.; Moreau, J.-F.; Déchanet-Merville, J. Antibody-dependent anti-cytomegalovirus activity of human γδ T cells expressing CD16 (FcγRIIIa). Blood 2012, 119, 1418–1427. [Google Scholar] [CrossRef]
- Gertner-Dardenne, J.; Bonnafous, C.; Bezombes, C.; Capietto, A.-H.; Scaglione, V.; Ingoure, S.; Cendron, D.; Gross, E.; Lepage, J.-F.; Quillet-Mary, A.; et al. Bromohydrin pyrophosphate enhances antibody-dependent cell-mediated cytotoxicity induced by therapeutic antibodies. Blood 2009, 113, 4875–4884. [Google Scholar] [CrossRef] [Green Version]
- Hoeres, T.; Pretscher, D.; Holzmann, E.; Smetak, M.; Birkmann, J.; Triebel, J.; Bertsch, T.; Wilhelm, M. Improving Immunotherapy Against B-Cell Malignancies Using γδ T-Cell-specific Stimulation and Therapeutic Monoclonal Antibodies. J. Immunother. 2019, 42, 331–344. [Google Scholar] [CrossRef]
- Zoine, J.T.; Knight, K.A.; Fleischer, L.C.; Sutton, K.S.; Goldsmith, K.C.; Doering, C.B.; Spencer, H.T. Ex vivo expanded patient-derived γδ T-cell immunotherapy enhances neuroblastoma tumor regression in a murine model. Oncoimmunology 2019, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Capietto, A.-H.; Martinet, L.; Fournié, J.-J. Stimulated γδ T cells increase the in vivo efficacy of trastuzumab in HER-2+ breast cancer. J. Immunol. 2011, 187, 1031–1038. [Google Scholar] [CrossRef] [Green Version]
- Oberg, H.H.; Kellner, C.; Gonnermann, D.; Sebens, S.; Bauerschlag, D.; Gramatzki, M.; Kabelitz, D.; Peipp, M.; Wesch, D. Tribody [(HER2)2xCD16] Is More Effective Than Trastuzumab in Enhancing γδ T Cell and Natural Killer Cell Cytotoxicity Against HER2-Expressing Cancer Cells. Front. Immunol. 2018, 9, 814. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Bioley, G.; Lévy, N.; Eberl, M.; Luo, M.; Tampé, R.; Lévy, F.; Romero, P.; Moser, B. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc. Natl. Acad. Sci. USA 2009, 106, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, L.J.; Paulis, L.E.; Tel, J.; Figdor, C.G. Towards efficient cancer immunotherapy: Advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014, 32, 456–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenmüller, W.; Kastenmüller, K.; Kurts, C.; Seder, R.A. Dendritic cell-targeted vaccines—Hope or hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar]
- Himoudi, N.; Morgenstern, D.A.; Yan, M.; Vernay, B.; Saraiva, L.; Wu, Y.; Cohen, C.J.; Gustafsson, K.; Anderson, J. Human γδ T lymphocytes are licensed for professional antigen presentation by interaction with opsonized target cells. J. Immunol. 2012, 188, 1708–1716. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Frencher, J.; Huang, D.; Wang, W.; Yang, E.; Chen, C.Y.; Zhang, Z.; Wang, R.; Qaqish, A.; Larsen, M.H.; et al. Immunization of Vγ2Vδ2 T cells programs sustained effector memory responses that control tuberculosis in nonhuman primates. Proc. Natl. Acad. Sci. USA 2019, 116, 6371–6378. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Huang, D.; Wei, H.; Wang, R.C.; Chen, C.Y.; Shen, L.; Zhang, W.; Jin, J.; Chen, Z.W. Expansion, reexpansion, and recall-like expansion of Vγ9Vδ2 T cells in smallpox vaccination and monkeypox virus infection. J. Virol. 2009, 83, 11959–11965. [Google Scholar] [CrossRef] [Green Version]
- Sicard, H.; Ingoure, S.; Luciani, B.; Serraz, C.; Fournié, J.-J.; Bonneville, M.; Tiollier, J.; Romagné, F. In vivo immunomanipulation of Vγ9Vδ2 T cells with a synthetic phosphoantigen in a preclinical nonhuman primate model. J. Immunol. 2005, 175, 5471–5480. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Kamath, A.; Das, H.; Li, L.; Bukowski, J.F. Antibacterial effect of human Vγ2Vδ2 T cells in vivo. J. Clin. Invest. 2001, 108, 1349–1357. [Google Scholar] [CrossRef]
- Huang, D.; Shen, Y.; Qiu, L.; Chen, C.Y.; Shen, L.; Estep, J.; Hunt, R.; Vasconcelos, D.; Du, G.; Aye, P.; et al. Immune Distribution and Localization of Phosphoantigen-Specific Vγ2Vδ2 T Cells in Lymphoid and Nonlymphoid Tissues in Mycobacterium tuberculosis Infection. Infect. Immun. 2008, 76, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, N.; Todaro, M.; Sireci, G.; Meraviglia, S.; Stassi, G.; Dieli, F. Mechanisms underlying lineage commitment and plasticity of human γδ T cells. Cell. Mol. Immunol. 2013, 10, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traxlmayr, M.W.; Wesch, D.; Dohnal, A.M.; Funovics, P.; Fischer, M.B.; Kabelitz, D.; Felzmann, T. Immune suppression by gammadelta T-cells as a potential regulatory mechanism after cancer vaccination with IL-12 secreting dendritic cells. J. Immunother. 2010, 33, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Oberg, H.-H.; Kabelitz, D.; Wesch, D. Phenotype and regulation of immunosuppressive Vδ2-expressing γδ T cells. Cell. Mol. Life Sci. 2013, 71, 1943–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, C.; Meyer, A.; Kouakanou, L.; Feder, J.; Schricker, T.; Lettau, M.; Janssen, O.; Wesch, D.; Kabelitz, D. TGF-β enhances the cytotoxic activity of Vδ2 T cells. Oncoimmunology 2019, 8, e1522471. [Google Scholar] [CrossRef] [Green Version]
- Wesch, D.; Peters, C.; Siegers, G.M. Human gamma delta T regulatory cells in cancer: Fact or fiction? Front. Immunol. 2014, 5, 598. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. γδT17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Ma, C.; Wang, F.; Hsueh, E.C.; Toth, K.; Huang, Y.; Mo, W.; Liu, S.; Han, B.; Varvares, M.A.; et al. Specific recruitment of γδ regulatory T cells in human breast cancer. Cancer Res. 2013, 73, 6137–6148. [Google Scholar] [CrossRef] [Green Version]
- Meraviglia, S.; Eberl, M.; Vermijlen, D.; Todaro, M.; Buccheri, S.; Cicero, G.; La Mendola, C.; Guggino, G.; D’Asaro, M.; Orlando, V.; et al. In vivo manipulation of Vγ9Vδ2 T cells with zoledronate and low-dose interleukin-2 for immunotherapy of advanced breast cancer patients. Clin. Exp. Immunol. 2010, 161, 290–297. [Google Scholar] [CrossRef]
- Dieli, F.; Vermijlen, D.; Fulfaro, F.; Caccamo, N.; Meraviglia, S.; Cicero, G.; Roberts, A.; Buccheri, S.; D’Asaro, M.; Gebbia, N.; et al. Targeting Human T Cells with Zoledronate and Interleukin-2 for Immunotherapy of Hormone-Refractory Prostate Cancer. Cancer Res. 2007, 67, 7450–7457. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, H.; Oberg, H.-H.; Juricke, M.; Marischen, L.; Kunz, M.; Mundhenke, C.; Gieseler, F.; Kabelitz, D.; Wesch, D. Toll-like receptors 3 and 7 agonists enhance tumor cell lysis by human gammadelta T cells. Cancer Res. 2009, 69, 8710–8717. [Google Scholar] [CrossRef] [Green Version]
- Wesch, D.; Beetz, S.; Oberg, H.-H.; Marget, M.; Krengel, K.; Kabelitz, D. Direct costimulatory effect of TLR3 ligand poly(I:C) on human gamma delta T lymphocytes. J. Immunol. 2006, 176, 1348–1354. [Google Scholar] [PubMed]
- Pietschmann, K.; Beetz, S.; Welte, S.; Martens, I.; Gruen, J.; Oberg, H.H.; Wesch, D.; Kabelitz, D. Toll-like receptor expression and function in subsets of human gammadelta T lymphocytes. Scand. J. Immunol. 2009, 70, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Wesch, D.; Peters, C.; Oberg, H.-H.; Pietschmann, K.; Kabelitz, D. Modulation of γδ T cell responses by TLR ligands. Cell. Mol. Life Sci. (Cmls) 2011, 68, 2357–2370. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Shao, L.; Wang, Y.; Chen, C.Y.; Huang, D.; Yao, S.; Zhan, X.; Sicard, H.; Wang, R.; Chen, Z.W. Phosphoantigen-activated V gamma 2V delta 2 T cells antagonize IL-2-induced CD4+CD25+Foxp3+ T regulatory cells in mycobacterial infection. Blood 2009, 113, 837–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo Presti, E.; Pizzolato, G.; Corsale, A.M.; Caccamo, N.; Sireci, G.; Dieli, F.; Meraviglia, S. γδ T Cells and Tumor Microenvironment: From Immunosurveillance to Tumor Evasion. Front. Immunol. 2018, 9, 1395. [Google Scholar] [CrossRef]
- Wu, D.; Wu, P.; Qiu, F.; Wei, Q.; Huang, J. Human γδT-cell subsets and their involvement in tumor immunity. Cell. Mol. Immunol. 2016, 14, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Wen, Q.; He, W.T.; Yang, J.H.; Zhou, C.Y.; Xiong, W.J.; Ma, L. Optimized protocols for γδ T cell expansion and lentiviral transduction. Mol. Med. Rep. 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rådestad, E.; Klynning, C.; Stikvoort, A.; Mogensen, O.; Nava, S.; Magalhaes, I.; Uhlin, M. Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. Oncoimmunology 2019, 8, e1535730. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Matsushita, H.; Hoshikawa, M.; Hasegawa, K.; Kokudo, N.; Kakimi, K. Adjuvant combination therapy with gemcitabine and autologous γδ T-cell transfer in patients with curatively resected pancreatic cancer. Cytotherapy 2017, 19, 473–485. [Google Scholar] [CrossRef]
- Kobayashi, H.; Tanaka, Y.; Yagi, J.; Osaka, Y.; Nakazawa, H.; Uchiyama, T.; Minato, N.; Toma, H. Safety profile and anti-tumor effects of adoptive immunotherapy using gamma-delta T cells against advanced renal cell carcinoma: A pilot study. Cancer Immunol. Immunother. 2006, 56, 469–476. [Google Scholar] [CrossRef]
- Kobayashi, H.; Tanaka, Y.; Yagi, J.; Minato, N.; Tanabe, K. Phase I/II study of adoptive transfer of γδ T cells in combination with zoledronic acid and IL-2 to patients with advanced renal cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Wada, I.; Matsushita, H.; Noji, S.; Mori, K.; Yamashita, H.; Nomura, S.; Shimizu, N.; Seto, Y.; Kakimi, K. Intraperitoneal injection of in vitro expanded Vγ9Vδ2 T cells together with zoledronate for the treatment of malignant ascites due to gastric cancer. Cancer Med 2014, 3, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Kondo, M.; Takahashi, T.; Fujieda, N.; Kondo, A.; Tamura, N.; Murakawa, T.; Nakajima, J.; Matsushita, H.; Kakimi, K. Ex vivo characterization of γδ T-cell repertoire in patients after adoptive transfer of Vγ9Vδ2 T cells expressing the interleukin-2 receptor β-chain and the common γ-chain. Cytotherapy 2013, 15, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Smetak, M.; Schaefer-Eckart, K.; Kimmel, B.; Birkmann, J.; Einsele, H.; Kunzmann, V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014, 12, 45. [Google Scholar] [CrossRef]
- Bennouna, J.; Bompas, E.; Neidhardt, E.M.; Rolland, F.; Philip, I.; Galéa, C.; Salot, S.; Saiagh, S.; Audrain, M.; Rimbert, M.; et al. Phase-I study of Innacell γδ™, an autologous cell-therapy product highly enriched in γ9δ2 T lymphocytes, in combination with IL-2, in patients with metastatic renal cell carcinoma. Cancer Immunol. Immunother. 2008, 57, 1599–1609. [Google Scholar] [CrossRef]
- Abe, Y.; Muto, M.; Nieda, M.; Nakagawa, Y.; Nicol, A.; Kaneko, T.; Goto, S.; Yokokawa, K.; Suzuki, K. Clinical and immunological evaluation of zoledronate-activated Vγ9 gamma delta T-cell-based immunotherapy for patients with multiple myeloma. Exp. Hematol. 2009, 37, 956–968. [Google Scholar] [CrossRef]
- Nakajima, J.; Murakawa, T.; Fukami, T.; Goto, S.; Kaneko, T.; Yoshida, Y.; Takamoto, S.; Kakimi, K. A phase I study of adoptive immunotherapy for recurrent non-small-cell lung cancer patients with autologous gamma delta T cells. Eur. J. Cardiothorac. Surg. 2010, 37, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Nicol, A.J.; Tokuyama, H.; Mattarollo, S.R.; Hagi, T.; Suzuki, K.; Yokokawa, K.; Nieda, M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br. J. Cancer 2011, 105, 778–786. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, A.; Kaneko, T.; Kamigaki, T.; Fujimoto, K.; Ozawa, M.; Saito, M.; Ariyoshi, N.; Goto, S. Zoledronate-activated Vγ9 γδ T cell-based immunotherapy is feasible and restores the impairment of γδ T cells in patients with solid tumors. Cytotherapy 2011, 13, 92–97. [Google Scholar] [CrossRef]
- Sakamoto, M.; Nakajima, J.; Murakawa, T.; Fukami, T.; Yoshida, Y.; Murayama, T.; Takamoto, S.; Matsushita, H.; Kakimi, K. Adoptive immunotherapy for advanced non-small cell lung cancer using zoledronate-expanded γδ Tcells: A phase I clinical study. J. Immunother. 2011, 34, 202–211. [Google Scholar] [CrossRef]
- Cui, J.; Wang, N.; Zhao, H.; Jin, H.; Wang, G.; Niu, C.; Terunuma, H.; He, H.; Li, W. Combination of radiofrequency ablation and sequential cellular immunotherapy improves progression-free survival for patients with hepatocellular carcinoma. Int. J. Cancer 2014, 134, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Kunzmann, V.; Eckstein, S.; Reimer, P.; Weissinger, F.; Ruediger, T.; Tony, H.-P. Gammadelta T cells for immune therapy of patients with lymphoid malignancies. Blood 2003, 102, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, J.M.; Kaikobad, M.R.; Wallace, M.; Staab, M.J.; Horvath, D.L.; Wilding, G.; Liu, G.; Eickhoff, J.C.; McNeel, D.G.; Malkovsky, M. Pilot trial of interleukin-2 and zoledronic acid to augment γδ T cells as treatment for patients with refractory renal cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 1447–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, L.J.N. Clinical applications of gamma delta T cells with multivalent immunity. Front. Immunol. 2014, 1–10. [Google Scholar]
- Bennouna, J.; Levy, V.; Sicard, H.; Senellart, H.; Audrain, M.; Hiret, S.; Rolland, F.; Bruzzoni-Giovanelli, H.; Rimbert, M.; Galéa, C.; et al. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vγ9Vδ2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol. Immunother. 2010, 59, 1521–1530. [Google Scholar] [CrossRef]
- Kunzmann, V.; Smetak, M.; Kimmel, B.; Weigang-Koehler, K.; Goebeler, M.; Birkmann, J.; Becker, J.; Schmidt-Wolf, I.G.H.; Einsele, H.; Wilhelm, M. Tumor-promoting versus tumor-antagonizing roles of γδ T cells in cancer immunotherapy: Results from a prospective phase I/II trial. J. Immunother. 2012, 35, 205–213. [Google Scholar] [CrossRef]
- Pressey, J.G.; Adams, J.; Harkins, L.; Kelly, D.; You, Z.; Lamb, L.S. In vivo expansion and activation of γδ T cells as immunotherapy for refractory neuroblastoma: A phase 1 study. Med. (Baltim. ) 2016, 95, e4909. [Google Scholar] [CrossRef]
- Brown, C.E.; Mackall, C.L. CAR T cell therapy: Inroads to response and resistance. Nat. Rev. Immunol. 2019, 19, 73–74. [Google Scholar] [CrossRef]
- Capsomidis, A.; Benthall, G.; Van Acker, H.H.; Fisher, J.; Kramer, A.M.; Abeln, Z.; Majani, Y.; Gileadi, T.; Wallace, R.; Gustafsson, K.; et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018, 26, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Benyamine, A.; Loncle, C.; Foucher, E.; Blazquez, J.-L.; Castanier, C.; Chrétien, A.-S.; Modesti, M.; Secq, V.; Chouaib, S.; Gironella, M.; et al. BTN3A is a prognosis marker and a promising target for Vγ9Vδ2 T cells based-immunotherapy in pancreatic ductal adenocarcinoma (PDAC). Oncoimmunology 2017, 0, 1–14. [Google Scholar] [CrossRef]
- Palakodeti, A.; Sandstrom, A.; Sundaresan, L.; Harly, C.; Nedellec, S.; Olive, D.; Scotet, E.; Bonneville, M.; Adams, E.J. The Molecular Basis for Modulation of Human Vγ9Vδ2 T Cell Responses by CD277/Butyrophilin-3 (BTN3A)-specific Antibodies. J. Biol. Chem. 2012, 287, 32780–32790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benyamine, A.; Le Roy, A.; Mamessier, E.; Gertner-Dardenne, J.; Castanier, C.; Orlanducci, F.; Pouyet, L.; Goubard, A.; Collette, Y.; Vey, N.; et al. BTN3A molecules considerably improve Vγ9Vδ2T cells-based immunotherapy in acute myeloid leukemia. Oncoimmunology 2016, 5, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gökbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete Hematologic and Molecular Response in Adult Patients With Relapsed/Refractory Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment With Blinatumomab: Results From a Phase II, Single-Arm, Multicenter Study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Yuraszeck, T.; Kasichayanula, S.; Benjamin, J.E. Translation and Clinical Development of Bispecific T-cell Engaging Antibodies for Cancer Treatment. Clin. Pharmacol. Ther. 2017, 101, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef]
- Klinger, M.; Brandl, C.; Zugmaier, G.; Hijazi, Y.; Bargou, R.C.; Topp, M.S.; Gökbuget, N.; Neumann, S.; Goebeler, M.; Viardot, A.; et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012, 119, 6226–6233. [Google Scholar] [CrossRef]
- Tang, J.; Pearce, L.; O’Donnell-Tormey, J.; Hubbard-Lucey, V.M. Trends in the global immuno-oncology landscape. Nat. Rev. Drug Discov. 2018, 17, 783–784. [Google Scholar] [CrossRef]
- Kebenko, M.; Goebeler, M.-E.; Wolf, M.; Hasenburg, A.; Seggewiss-Bernhardt, R.; Ritter, B.; Rautenberg, B.; Atanackovic, D.; Kratzer, A.; Rottman, J.B.; et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology 2018, 7, e1450710. [Google Scholar]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Yang, T.-H.O.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Kobold, S.; Pantelyushin, S.; Rataj, F.; Vom Berg, J. Rationale for Combining Bispecific T Cell Activating Antibodies with Checkpoint Blockade for Cancer Therapy. Front. Oncol. 2018, 8, 285. [Google Scholar] [CrossRef]
- Marshall, H.T.; Djamgoz, M.B.A. Immuno-Oncology: Emerging Targets and Combination Therapies. Front. Oncol. 2018, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Dopfer, E.P.; Hartl, F.A.; Oberg, H.-H.; Siegers, G.M.; Yousefi, O.S.; Kock, S.; Fiala, G.J.; Garcillán, B.; Sandstrom, A.; Alarcón, B.; et al. The CD3 conformational change in the γδ T Cell Receptor is not triggered by antigens but can be enforced to enhance tumor killing. CellReports 2014, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, O.; Wesselborg, S.; Heckl-Ostreicher, B.; Pechhold, K.; Bender, A.; Schondelmaier, S.; Moldenhauer, G.; Kabelitz, D. T cell receptor/CD3-signaling induces death by apoptosis in human T cell receptor gamma delta + T cells. J. Immunol. 1991, 146, 35–39. [Google Scholar] [PubMed]
- Oberg, H.H.; Peipp, M.; Kellner, C.; Sebens, S.; Krause, S.; Petrick, D.; Adam-Klages, S.; Rocken, C.; Becker, T.; Vogel, I.; et al. Novel Bispecific Antibodies Increase T-Cell Cytotoxicity against Pancreatic Cancer Cells. Cancer Res. 2014, 74, 1349–1360. [Google Scholar] [CrossRef] [Green Version]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. CANCER IMMUNOLOGY. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef]
- Oberg, H.-H.; Kellner, C.; Peipp, M.; Sebens, S.; Adam-Klages, S.; Gramatzki, M.; Kabelitz, D.; Wesch, D. Monitoring Circulating γδ T Cells in Cancer Patients to Optimize γδ T Cell-Based Immunotherapy. Front. Immunol. 2014, 5, 643. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Cesano, A.; Warren, S. Bringing the next Generation of Immuno-Oncology Biomarkers to the Clinic. Biomedicines 2018, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Duell, J.; Dittrich, M.; Bedke, T.; Mueller, T.; Eisele, F.; Rosenwald, A.; Rasche, L.; Hartmann, E.; Dandekar, T.; Einsele, H.; et al. Frequency of regulatory T cells determines the outcome of the T-cell-engaging antibody blinatumomab in patients with B-precursor ALL. Leukemia 2017, 31, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. CELL 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holderness, J.; Hedges, J.F.; Ramstead, A.; Jutila, M.A. Comparative Biology of γδ T Cell Function in Humans, Mice, and Domestic Animals. Annu. Rev. Anim. Biosci. 2013, 1, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.M.; Winkler, T.H. Resolving the mystery—How TCR transgenic mouse models shed light on the elusive case of gamma delta T cells. J. Leukoc. Biol. 2020, 154, 5821–16. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Man, F.; Lim, L.; Volpe, A.; Gabizon, A.; Shmeeda, H.; Draper, B.; Parente-Pereira, A.C.; Maher, J.; Blower, P.J.; Fruhwirth, G.O.; et al. In Vivo PET Tracking of 89Zr-Labeled Vγ9Vδ2 T Cells to Mouse Xenograft Breast Tumors Activated with Liposomal Alendronate. Mol. Ther. 2019, 27, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Kabelitz, D.; Wesch, D.; Pitters, E.; Zöller, M. Characterization of Tumor Reactivity of Human Vγ9Vδ2 γδ T Cells In Vitro and in SCID Mice In Vivo. J. Immunol. 2004, 173, 6767–6776. [Google Scholar] [CrossRef] [Green Version]
- Chitadze, G.; Oberg, H.-H.; Wesch, D.; Kabelitz, D. The Ambiguous Role of γδ T Lymphocytes in Antitumor Immunity. Trends Immunol. 2017, 38, 668–678. [Google Scholar] [CrossRef]
- Jarry, U.; Chauvin, C.; Joalland, N.; Léger, A.; Minault, S.; Robard, M.; Bonneville, M.; Oliver, L.; Vallette, F.M.; Vié, H.; et al. Stereotaxic administrations of allogeneic human Vγ9Vδ2 T cells efficiently control the development of human glioblastoma brain tumors. Oncoimmunology 2016, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mao, T.; Miao, C.; Liao, Y.; Chen, Y.; Yeh, C.; Liu, C. Ex Vivo Expanded Human Vγ9Vδ2 T-Cells Can Suppress Epithelial Ovarian Cancer Cell Growth. Int. J. Mol. Sci. 2019, 20, 1139. [Google Scholar] [CrossRef] [Green Version]
- Santolaria, T.; Robard, M.; Léger, A.; Catros, V.; Bonneville, M.; Scotet, E. Repeated Systemic Administrations of Both Aminobisphosphonates and Human Vγ9Vδ2 T Cells Efficiently Control Tumor Development In Vivo. J. Immunol. 2013, 191, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.J.; Chan, K.W.; Im, S.; Chua, D.; Sham, J.S.; Tin, P.C.; He, Z.M.; Ng, M.H. Anti-tumor effects of human peripheral gammadelta T cells in a mouse tumor model. Int. J. Cancer 2001, 92, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, E.; Bocca, P.; Emionite, L.; Cilli, M.; Cipollone, G.; Morandi, F.; Raffaghello, L.; Pistoia, V.; Prigione, I. Mechanisms of the Antitumor Activity of Human Vγ9Vδ2 T Cells in Combination With Zoledronic Acid in a Preclinical Model of Neuroblastoma. Mol. Ther. 2013, 21, 1034–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgins, N.O.; Al-Jamal, W.T.; Wang, J.T.W.; Klippstein, R.; Costa, P.M.; Sosabowski, J.K.; Marshall, J.F.; Maher, J.; Al-Jamal, K.T. Investigating in vitro and in vivo αvβ6 integrin receptor-targeting liposomal alendronate for combinatory γδ T cell immunotherapy. J. Control. Release 2017, 256, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Parente-Pereira, A.C.; Shmeeda, H.; Whilding, L.M.; Zambirinis, C.P.; Foster, J.; van der Stegen, S.J.C.; Beatson, R.; Zabinski, T.; Brewig, N.; Sosabowski, J.K.; et al. Adoptive Immunotherapy of Epithelial Ovarian Cancer with Vγ9Vδ2 T Cells, Potentiated by Liposomal Alendronic Acid. J. Immunol. 2014, 193, 5557–5566. [Google Scholar] [CrossRef] [Green Version]
- Gertner-Dardenne, J.; Castellano, R.; Mamessier, E.; Garbit, S.; Kochbati, E.; Etienne, A.; Charbonnier, A.; Collette, Y.; Vey, N.; Olive, D. Human Vγ9Vδ2 T Cells Specifically Recognize and Kill Acute Myeloid Leukemic Blasts. J. Immunol. 2012, 188, 4701–4708. [Google Scholar] [CrossRef]
- Malkovska, V.; Cigel, F.K.; Armstrong, N.; Storer, B.E.; Hong, R. Antilymphoma activity of human gamma delta T-cells in mice with severe combined immune deficiency. Cancer Res. 1992, 52, 5610–5616. [Google Scholar]
- Xiang, Z.; Liu, Y.; Zheng, J.; Liu, M.; Lv, A.; Gao, Y.; Hu, H.; Lam, K.-T.; Chan, G.C.-F.; Yang, Y.; et al. Targeted Activation of Human Vγ9Vδ2 T Cells Controls Epstein-Barr Virus-Induced B Cell Lymphoproliferative Disease. Cancer Cell 2014, 26, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Zumwalde, N.A.; Sharma, A.; Xu, X.; Ma, S.; Schneider, C.L.; Romero-Masters, J.C.; Hudson, A.W.; Gendron-Fitzpatrick, A.; Kenney, S.C.; Gumperz, J.E. Adoptively transferred Vγ9Vδ2 T cells show potent antitumor effects in a preclinical B cell lymphomagenesis model. J. Clinic. Invest. Insight 2017, 2, 3202–3215. [Google Scholar] [CrossRef] [Green Version]
- Pechhold, K.; Wesch, D.; Schondelmaier, S.; Kabelitz, D. Primary activation of Vγ9-expressing gamma delta T cells by Mycobacterium tuberculosis. Requirement for Th1-type CD4 T cell help and inhibition by IL-10. J. Immunol. 1994, 152, 4984–4992. [Google Scholar]
- Chen, J.; Niu, H.; He, W.; Ba, D. Antitumor activity of expanded human tumor-infiltrating gammadelta T lymphocytes. Int. Arch. Allergy Immunol. 2001, 125, 256–263. [Google Scholar] [CrossRef]
- D’Asaro, M.; La Mendola, C.; Di Liberto, D.; Orlando, V.; Todaro, M.; Spina, M.; Guggino, G.; Meraviglia, S.; Caccamo, N.; Messina, A.; et al. Vγ9Vδ2 T Lymphocytes Efficiently Recognize and Kill Zoledronate-Sensitized, Imatinib-Sensitive, and Imatinib-Resistant Chronic Myelogenous Leukemia Cells. J. Immunol. 2010, 184, 3260–3268. [Google Scholar] [CrossRef] [Green Version]
- Beck, B.H.; Kim, H.-G.; Kim, H.; Samuel, S.; Liu, Z.; Shrestha, R.; Haines, H.; Zinn, K.; Lopez, R.D. Adoptively transferred ex vivo expanded gammadelta-T cells mediate in vivo antitumor activity in preclinical mouse models of breast cancer. Breast Cancer Res. Treat. 2010, 122, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokouhaki, P.; Han, M.; Joe, B.; Li, M.; Johnston, M.R.; Tsao, M.-S.; Zhang, L. Adoptive immunotherapy of cancer using ex vivo expanded human γδ T cells: A new approach. Cancer Lett. 2010, 297, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, F.; Pende, D.; Burgio, V.L.; Castelli, C.; Spada, M.; Venditti, M.; Luciani, F.; Lugini, L.; Federici, C.; Ramoni, C.; et al. Effect of human natural killer and gammadelta T cells on the growth of human autologous melanoma xenografts in SCID mice. Cancer Res. 2004, 64, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberg, H.-H.; Peters, C.; Kabelitz, D.; Wesch, D. Real-time cell analysis (RTCA) to measure killer cell activity against adherent tumor cells in vitro. Meth. Enzym. 2020, 631, 429–441. [Google Scholar]
- García-Martínez, E.; Smith, M.; Buqué, A.; Aranda, F.; de la Peña, F.A.; Ivars, A.; Cánovas, M.S.; Conesa, M.A.V.; Fucikova, J.; Spisek, R.; et al. Trial Watch: Immunostimulation with recombinant cytokines for cancer therapy. Oncoimmunology 2018, 7, 1–16. [Google Scholar]
- Starick, L.; Riaño, F.; Karunakaran, M.M.; Kunzmann, V.; Li, J.; Kreiss, M.; Amslinger, S.; Scotet, E.; Olive, D.; De Libero, G.; et al. Butyrophilin 3A (BTN3A, CD277)-specific antibody 20.1 differentially activates Vγ9Vδ2 TCR clonotypes and interferes with phosphoantigen activation. Eur. J. Immunol. 2017, 47, 982–992. [Google Scholar] [CrossRef]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef]


| Reference | Indication | Treatment | Ex Vivo Expansion Stimulus | n | Response |
|---|---|---|---|---|---|
| [100] | RCC | γδ T cells | 2M3B1-PP + IL-2 Teceleukin | 7 | 3 PR |
| [105] | RCC | Innacell γδ T cells + IL-2 | BrHPP1 + IL-2 Proleukin | 10 | 6 SD |
| [106] | MM | γδ T cells | Zoledronate + IL-2 | 6 | 0 |
| [107] | NSCLC | γδ T cells | Zoledronate + IL-2 | 10 | 3 SD |
| [101] | RCC | γδ T cells + Zoledronate + IL-2 | 2M3B1-PP | 11 | 1 CR 5 SD |
| [108] | Diverse solid tumors | γδ T cells + Zoledronate | Zoledronate + IL-2 | 18 | 3 SD |
| [109] | Diverse solid tumors | γδ T cells + combinations | Zoledronate + IL-2 | 25 | 3 PR |
| [110] | NSCLC | γδ T cells | Zoledronate + IL-2 | 15 | 6 SD |
| [111] | HCC | Radiofreqency ablation + cytokines | NK2, CIK3, γδ T stimuli | 30 | |
| [103] | CRC | γδ T cells | Zoledronate + IL-2 | 6 | |
| [102] | Gastric cancer | γδ T cells + Zoledronate | Zoledronate + IL-2 | 7 | 1 PR, 1 CR |
| [99] | Pancreatic Cancer | γδ T cells + Gemcitabine | Zoledronate + IL-2 | 28 | |
| [104] | Hematological | γδ T cells (family donor) Zoledronate + IL-2 | CD4+ and CD8+ T cell depleted PBMCs | 4 | 3 CR |
| Reference | Indication | Treatment | n= | Response | Response Biomarker |
|---|---|---|---|---|---|
| [112] | Hematological (NHL + MM) | Pamidronate + IL-2 | 19 | 3 SDs | Vγ9Vδ2 PBL |
| [89] | Prostate Cancer | Zoledronate/Zoledronate + IL-2 | 18 | 1 SD, 1 PR 4 SD, 2 PR | TRAIL, Vγ9Vδ2 PBL |
| [115] | RCC, CRC, Breast Cancer | BrHPP + IL-2 | 28 | ||
| [88] | Breast Cancer | Zoledronate + IL-2 | 10 | 2 SD, 1 PR | Vγ9Vδ2 PBL |
| [113] | Metastatic RCC | Zoledronate + IL-2 | 12 | Vγ9Vδ2 PBL | |
| [116] | RCC, melanoma, AML | Zoledronate + IL-2 | 21 | 0 in solid tumors, 2 PR in AML | IFN-γ and in vivo expansion |
| [117] | Refractory neuroblastoma | Zoledronate + IL-2 | 4 | 1 SD | Vγ9Vδ2 PBL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Künkele, K.-P.; Wesch, D.; Oberg, H.-H.; Aichinger, M.; Supper, V.; Baumann, C. Vγ9Vδ2 T Cells: Can We Re-Purpose a Potent Anti-Infection Mechanism for Cancer Therapy? Cells 2020, 9, 829. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040829
Künkele K-P, Wesch D, Oberg H-H, Aichinger M, Supper V, Baumann C. Vγ9Vδ2 T Cells: Can We Re-Purpose a Potent Anti-Infection Mechanism for Cancer Therapy? Cells. 2020; 9(4):829. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040829
Chicago/Turabian StyleKünkele, Klaus-Peter, Daniela Wesch, Hans-Heinrich Oberg, Martin Aichinger, Verena Supper, and Christoph Baumann. 2020. "Vγ9Vδ2 T Cells: Can We Re-Purpose a Potent Anti-Infection Mechanism for Cancer Therapy?" Cells 9, no. 4: 829. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9040829