Modulation of mTORC1 Signaling Pathway by HIV-1




1
CNRS UMR 9018, Université Paris-Saclay, Institut Gustave Roussy, 114, rue Édouard Vaillant, 94805 Villejuif, France
2
LIA 1066 LFR2O French-Russian Joint Cancer Research Laboratory, 114, rue Édouard-Vaillant, 94805 Villejuif, France
3
Koltzov Institute of Developmental Biology, 26, Vavilova str., 119334 Moscow, Russia
*
Author to whom correspondence should be addressed.
Cells 2020, 9(5), 1090; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051090
Submission received: 25 March 2020
/
Revised: 24 April 2020
/
Accepted: 25 April 2020
/
Published: 28 April 2020
(This article belongs to the Collection Compartmentilisation of Cellular Signaling)
Abstract
:Mammalian target of rapamycin complex 1 (mTORC1) is a master regulator of cellular proliferation and survival which controls cellular response to different stresses, including viral infection. HIV-1 interferes with the mTORC1 pathway at every stage of infection. At the same time, the host cells rely on the mTORC1 pathway and autophagy to fight against virus replication and transmission. In this review, we will provide the most up-to-date picture of the role of the mTORC1 pathway in the HIV-1 life cycle, latency and HIV-related diseases. We will also provide an overview of recent trends in the targeting of the mTORC1 pathway as a promising strategy for HIV-1 eradication.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Viruses generate an environment that is favorable for their successful replication and transmission during infection. In order to optimize their biosynthetic needs, viruses use the nutrient, energy and macromolecule synthesis systems of the host cells and manipulate their metabolism [1]. Host cells in turn respond to viral infection by changing their transcriptional and translational programs and employing antiviral metabolic changes [2,3,4]. Cellular response to various stresses, including viral infection, is under the control of the mechanistic target of rapamycin complex 1 (mTORC1), which drives proliferation and survival by the regulation of anabolic and catabolic processes. Thus, it is no wonder that viruses try to use this signaling pathway to their benefit [4].
The human immunodeficiency virus type-1 (HIV-1) is a lentivirus containing two positive-sense single strand RNAs encapsulated in a capsid formed by p24. Structural HIV-1 proteins (Gag, Pol and Env) are produced as polypeptides and subsequently processed into matrix proteins, protease, reverse transcriptase, integrase and surface proteins gp120 and gp41. HIV-1 also codes for two regulatory components: Tat (transcriptional trans-activator) and Rev (regulator of expression of virion proteins). Finally, Vpr, Vif, Nef and Vpu serve as accessory regulatory elements [5]. During viral entry, gp120 binds to the CD4 molecule of the host cell and gp41 binds to the cellular coreceptors such as CCR5 and CXCR4. After fusion with the host cell, a conical capisid around the HIV-1 genome disassembles (a process known as uncoating), and viral RNA is released into the cytoplasm where it is transcribed by a viral-encoded reverse transcriptase. Uncoating probably occurs in the cytoplasm in coordination with reverse transcription or at the nuclear envelope during nuclear import. Subsequently, viral dsDNA uses the host nuclear import machinery to move to the host cell nucleus, where it integrates into the host DNA with the help of a viral-encoded integrase. Remarkably, recent studies revealed that intact viral cores can enter to the nucleus and uncoat just before integration to their chromosomal integration sites [6]. Pro-viruses use the host RNA polymerase to synthetize mRNA, which is subsequently translated into viral proteins. HIV-1 infects and kills cells of the immune system such as T-helper cells, macrophages and dendritic cells, leading to immunodeficiency and further increasing the incidence of opportunistic infections and cancers.
The mechanistic target of rapamycin (mTOR) is an evolutionarily-conserved, serine-threonine protein kinase that belongs to the phosphatidylinositol 3-kinase PI3K-related family. mTOR forms two different macromolecular protein complexes, mTORC1 and mTORC2, which differ in their composition, downstream targets and regulation [7]. mTORC1 is sensitive, while mTORC2 is much less responsive to an allosteric mTOR inhibitor rapamycin (Sirolimus®), an immunosuppressor, which suppresses T and B cell activation by inhibition of the cell cycle. Various analogues of rapamycin, so called rapalogues (Everolimus®, Temsirolimus®), are also frequently used in clinics for immunosuppression. In addition, a number of alternative mTOR inhibitors have been developed. These inhibitors block both mTORC1 and mTORC2 (pan-inhibitors or TOR-KIs, i.e., INK128) or act on mTOR kinase and another protein (dual inhibitors), most often targeting a network upstream of mTORC1/2 [8]. Viruses are the leading cause of infections after solid-organ transplant and during anticancer treatment; the use of mTOR inhibitors decreases the incidence of viral infection in these medical conditions [9,10,11].
One of the first pieces of evidence that mTORC1 was involved in HIV-1 infection came from the observation that treatment with rapamycin causes downregulation of CCR5 expression in T cells [12]. A number of studies that immediately followed confirmed that rapamycin possessed anti-HIV-1 properties both in vitro and in vivo, pointing to the mTORC1 importance during HIV-1 propagation (reviewed in [13,14]). Pan-inhibitors of mTORC1 block HIV-1 even more efficiently, interfering both with virus entry (by reducing CCR5 levels) and with basal and induced transcription, as shown in preclinical humanized mice models [15]. Our review is focused on recently discovered mechanisms of mTORC1 contribution to HIV-1 infection, latency and development of HIV-1 related diseases.
2. mTORC1, a Main Metabolic Network of the Cell
mTORC1 integrates signals from many intracellular and extracellular cues: amino acids, growth factors, energy, oxygen, DNA damage and infectious agents, including viruses. Depending on the nature of the signal, its duration, cell type and many other factors, mTORC1 will “determine” the subsequent cell fate. mTORC1 can accelerate proliferation via the phosphorylation of its key targets, p70S6 Kinase 1 (S6K1) and members of eIF4E Binding Protein family (4E-BPs), which participate in the synthesis of three major cell constituents: proteins, nucleotides and lipids [7]. Alternatively, mTORC1 can drive the cell through a catabolic process, since it controls both autophagy (via phosphorylation of ULK1 and TFEB) and protein degradation by ubiquitin-proteasome system (via phosphorylation of ERK5) [16]. In order to operate through signals and responses, mTORC1 manipulates one of the most complex signaling networks in the cell, the mTORC1 pathway.
Growth factors and cytokines activate mTORC1 through the PI3K-AKT signaling pathway [17]. Signals from these stimuli activate serine-threonine kinase AKT, leading to the phosphorylation and inactivation of the TSC2 component of tuberous sclerosis complex (TSC), an mTORC1 inhibitor. TSC inactivation results in the stimulation of RHEB GTPase, which activates mTORC1 [18] (Figure 1). AKT can also activate mTORC1 directly, in a TSC-independent way, by phosphorylating and inactivating the mTORC1 inhibitory component, PRAS40 [19]. Amino acids activate mTORC1 through a number of specific amino acid sensors. These sensors in turn stimulate GATOR1/GATOR2 complexes, containing a number of proteins, including tumor suppressors such as DEPDC5 [20]. Stimulation of GATORs further induces Rag GTPases (RagA/B and RagC/D), leading to mTORC1 activation (Figure 1) [21]. In contrast, low energy levels and hypoxic conditions result in mTORC1 inhibition via stimulation of AMP-activated protein kinase (AMPK) [22,23]. DNA damaging agents also suppress mTORC1 activity, in part by activation of the most powerful mTORC1 antagonist, p53 (Figure 1) [8,24].
Viruses can both activate and inhibit the mTORC1 pathway, depending on the type of the virus, stage on infection and cells in contact with the virus (reviewed in [4]). HIV-1 infection is generally associated with inhibition of mTORC1 in CD4 T cells at the early stage of infection and the activation of mTORC1 at the late stage (see below). This behavior is often related with the ability of mTORC1 to inhibit autophagy. Indeed, at the early state of infection, host cells try to activate autophagy and/or apoptosis to eliminate the spread of the virus; therefore, they need to suppress mTORC1 activity. In contrast, at the late state of infection, when host cells have “lost the battle”, viruses try to activate mTORC1 in order to suppress autophagy. Interestingly, viruses can target the mTORC1 pathway both upstream and downstream of mTORC1. For instance, Kaposi’s sarcoma-associated herpes virus (KSHV), related to the AIDS defining cancer of the same name, activates mTORC1 upstream through the PI3K-AKT axis [25], while herpesviruses can also act downstream of the mTORC1 node, mainly through stimulation of phosphorylation and inactivation of 4E-BP1 (Figure 1) [4].
3. mTORC1 in the Immune System
mTORC1 plays a central role in the metabolism, differentiation and effector functions of immune cells [26,27]. Here, we will only briefly describe the role of mTORC1 in B and T lymphocytes, as they are the most relevant during HIV-1 infection and in the development of HIV-1-related diseases (e.g., B-cell lymphomas). Many more details can be found in exhaustive recent reviews about the role of mTORC1 in B cells [28], T cells [29] and NK, macrophages and dendritic cells [30].
T lymphocytes in the resting state are catabolic and use autophagy to produce molecules necessary for protein synthesis and energy. After activation, T cells become anabolic and switch to glycolysis to produce energy and various substrates for proliferation. A transition from the resting to active state requires the upregulation of metabolic pathways controlled by mTORC1.
CD4 T cells, which are the primary target of HIV-1, are effector T cells which can be differentiated into distinct effector lineages (Th1, Th2, Th17) in response to various inflammatory cytokines. mTORC1 helps this differentiation process by promoting glycolysis and lipid biosynthesis [31,32,33,34]. mTORC1-deficient T cells have an impaired ability to differentiate [31]. Similarly, T cells lacking the mTORC1 activator RHEB also fail to differentiate into Th1 and Th17 [32]. Alternatively, the deletion of mTORC1 inhibitor TSC1 results in enhanced mTORC1 activity, leading to elevated Th1 and Th17 differentiation and multiorgan inflammation in mice [35]. In addition, a deficiency in the leucine transporter LAT1 [36] and glutamine transporter ASCT2 [37] impairs Th1 and Th17 differentiation in a mTORC1-dependent manner.
CD8 T cells have the ability to kill cancer cells or virally-infected cells (effector cytotoxic T cells), and maintain long-term memory (memory T cells). Increased mTORC1 activity is beneficial for the generation of effector CD8 T cells. In contrast, active mTORC1 downregulates memory T cell formation, resulting in a decreased response to secondary immunization [38,39,40].
Tregs are immune cells that negatively control both cytotoxic effector CD8 T cells and effector CD4 T cells. mTORC1 activity has negative effects on Treg generation [41], while it is positively correlated with the inhibitory functions of Tregs towards other T cells [42]. Active mTORC1 promotes the conversion of Tregs to effector-like T cells and further impairs Treg stability and function [35,43,44]. Toll-like receptor signals, which promote Treg proliferation, increase mTORC1 signaling, glycolysis and expression of glucose transporter Glut1 [43].
B cells undergo multiple steps of maturation before becoming plasma cells and acquiring antibody-secreting capacity, which is critical for defense against recurrent infections. mTORC1 is highly active at the earliest stages of B cell development, and its activity reduces during B cell maturation [45,46,47]. Homozygous deletion of mTORC1 component RAPTOR prior to lineage specification results in a total lack of B cell maturation [45,46].
Thus, mTORC1 senses both intra- and extra- cellular cues, but also the immune microenvironment, in order to influence the differentiation and maturation of immune cells.
4. HIV-1 Modulates mTORC1 Activity
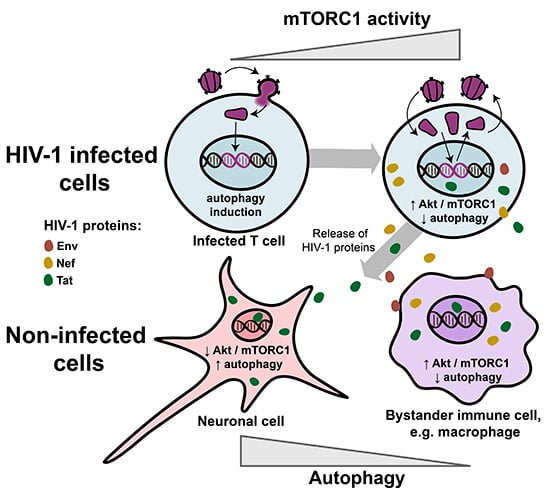
HIV-1 infection, similarly to other viral infections, generally increases mTORC1 activity both in productively-infected and bystander host cells, thus promoting successful viral integration and replication [48,49,50,51] (graphical abstract). Moreover, mTORC1 activity is required for optimal synthesis of viral proteins, e.g., Gag [52]. mTORC1 is also activated following HIV-1 infection in peripheral blood mononuclear cells and in several model cell lines such as Jurkat cells, HeLa and HEK293 [48,49,52,53]. Nevertheless, HIV-1 is not able to sustain mTORC1 activity in conditions of nutrient deprivation or upon pharmacological inhibition of mTORC1.
When amino acids are present, GTP-loaded RagA and RagB induce mTORC1 translocation from the cytoplasm to late endosomes/lysosomes (LELs) [7]. Interestingly, Gag can also be found at late endosomes during HIV-1 egress [54]. Nutrient starvation or oxidative stress result in the accumulation of a pool of mTOR-associated LELs at a juxtanuclear position. HIV-1 is capable of redistributing perinuclear clusters of mTORC1-associated LELs [52]. This HIV-1 activity depends on the RagA and RagB, which can interact with Vif and Gag, although nothing is known about the molecular mechanisms of these interactions. Depletion of RagA and RagB reduces virus production and results in the accumulation of viral particles at the plasma membrane [52].
Although HIV-1 does not enter or replicate in neurons, its infection of brain glia cells can induce neurotoxicity and inflammation. mTORC1 activation is involved in the interaction between the proinflammatory extracellular matrix protein osteopontin in the brain and HIV-1 Env proteins, which stimulates neurite growth [55].
mTORC1 activity can be modulated both by the entire HIV-1 and by its different proteins (Figure 2). For example, mTORC1 can be activated by treatment with HIV-1 Nef, Env or Tat proteins, resulting in efficient viral replication and the generation of new virions [48,49,51,56]. In contrast, combined treatment of neuronal cells with HIV-1 Tat and methamphetamine (a stimulant drug which increases exposure to HIV-1) results in the inhibition of mTORC1 activity [57]. As Tat is present in high concentrations in patient blood serum [58,59], and because of Tat’s ability to enter almost any cell in the human body due to its cell penetration domain [60], this viral transactivator can probably remotely regulate mTORC1 activity in many noninfected cells.
5. Regulation of Autophagy by HIV-1
mTORC1 is the negative regulator of the major cellular catabolic process, i.e., autophagy [61,62]. Autophagy plays a critical role in maintaining cellular homeostasis, but is also related to many pathological conditions including cancer and neurodegeneration [63]. Recent studies revealed an important role of autophagy in the context of HIV-1 infection in T cells, macrophages, dendritic cells and neuronal cells [64,65] (graphical abstract, Figure 2).
Autophagy can play both pro- or anti- viral roles, depending on the stage of HIV-1 infection and the cells in contact with virus and its components. HIV-1 requires early, nondegradative autophagic events for its replication, probably because the autophagosomal membrane provides a scaffold for virus assembly [66]. The entire virus and its proteins, in particular Tat, Nef and Env, can induce autophagy in infected cells to maximize HIV-1 production. At the same time, the virus has developed multiple strategies to escape the degradation of newly-synthetized viral proteins [64]. For example, Nef can interact with BECLIN-1, an autophagy initiation protein, to inhibit autophagosome maturation in macrophages and T cells [66,67,68,69]. Nef/BECLIN-1 interaction inhibits autophagy at the transcriptional level by preventing nuclear translocation of the pro-autophagic factor TFEB in an mTORC1-dependent manner [67]. Nef can also block autophagosome formation by increasing the interaction between BECLIN-1 and its inhibitor BCL2, an interaction that requires E3-ubiquitin ligase PRKN [68]. HIV-1 proteins can also disrupt autophagy in uninfected cells. For example, HIV-1 Tat inhibits autophagy induction in noninfected primary macrophages through AKT activation [50].
At the initial steps of infection, HIV-1 envelope proteins at the surface of the virus bind to CD4 receptors, mainly CCR5, initiating autophagy in CD4 T cells [70]. This autophagic process represents an anti-HIV response of the host cell, because it selectively degrades HIV-1 Tat [71,72]. Similarly, the induction of autophagy in macrophages with dual inhibitors of PI3K and mTORC1 results in the degradation of intracellular viral particles and the reduction of viral release [73]. The virus has, however, evolved to counteract autophagy [72]. Different HIV-1 proteins participate in this inhibition in different cells and at various steps of infection. For example, Vpr tries to inhibit autophagy in T cells already at the early stages of infection to facilitate efficient viral replication [74]; Vif inhibits autophagy at the late steps of viral replication in CD4+ T cells [75]; Nef blocks autophagy in macrophages and in T cells [67,68]; HIV-1 Tat blocks autophagy in bystander macrophages [50], while envelope proteins inhibit autophagy through mTORC1 activation in dendritic cells, but not in macrophages and CD4+ T cells [56].
The regulation of autophagy was also studied in the context of HIV-1 infection in neuronal cells, as HIV-1-associated neurocognitive disorders lead to increased mortality in HIV-1 patients [76]. HIV-1 Tat induces autophagy and increases neurotoxicity [77,78,79]. HIV-1 Tat can even trigger neuronal cell death when combined with methamphetamine [57,80,81]. HIV-1 Tat also plays a role in the regulation of mitophagy, a specialized form of autophagy, which removes damaged mitochondria. Treatment with HIV-1 Tat of primary microglial cells, the key target for HIV-1 infection in the central nervous system, results in the accumulation of damaged mitochondria and increased expression of mitophagy-signaling proteins PINK1 and PARKIN. However, even though mitophagy was induced, mitophagosomes fail to fuse with lysosomes and accumulate in the cells, resulting in overall defective mitophagy and subsequent neuroinflamation [82].
6. mTORC1 in HIV-1 Latency
Combination antiretroviral therapy (cART), which efficiently controls viral replication, remarkably improves the quality of life of HIV-1-positive individuals, although this therapy is not available to all people living with HIV-1, particularly in countries with limited resources. In addition, elevated risks of chronic inflammation [83], asynchronous muscle aging [84], the increased occurrence of various cancers [85,86] and developing HIV-1 drug resistance [87,88] create an urgent need to find a cure rather than to control of HIV-1 replication.
One of the major problems preventing the complete eradication of HIV-1 is the existence of latent reservoirs, “a cell type or anatomical site in which replication-competent form of virus accumulates and persists in spite of long periods of ART-suppression viremia” [89,90]. HIV-1 cellular reservoirs consist of resting memory CD4+ T cells, myeloid cells, macrophages and dendritic cells (DC) which allow the virus to survive and replicate [91]. Although the existence of anatomical HIV-1 reservoirs remains debated, the lymphoid tissues (spleen, thymus, lymph nodes and gut-associated lymphoid tissues), which are the most important sites of viral replication during infection, are considered the most prominent reservoirs.
Two opposite strategies are proposed to disrupt HIV-1 latency: “shock and kill” and “block and lock” (deep latency) [92,93]. In the “shock and kill” approach, the latent HIV-1 is reactivated (“shock”) by latency reversal agents (LRAs). The re-activated cells are further “killed” either by the attack of the host immune system or by the cytotoxic effect of HIV-1 itself, while uninfected cells are protected against viral infection by cART. Unfortunately, none of the LRAs tested so far have shown a significant effect on the viral reservoir in clinical trials [94]. In addition, agents inducing global T cell activation induced severe adverse reactions, mainly through inflammation, and were abandoned after several clinical trials. “Shocking” alone seems to be insufficient to eliminate latent T-cell reservoir. Effective “killing” strategies need to be developed to optimize this approach. An alternative “block and lock” strategy aims to bring the proviral HIV-1 to a deeply silenced state so that HIV-1 will not rebound, even if cART is discontinued. Recent findings demonstrate that manipulation of the mTORC1 pathway is important for the efficient development of both strategies.
The first notion of the importance of mTORC1 in HIV-1 latency came from the study by Besnard et al., who demonstrated that both mTORC1 and mTORC2 complexes are essential for HIV-1 reactivation from latency [95]. Accordingly, inhibitors which block both mTORC1 and mTORC2, pp242 and Torin1, prevent HIV-1 reactivation in primary CD4+ T cells from uninfected donors when infected ex vivo [49,95]. Rapamycin, which mostly blocks mTORC1, is a less effective inhibitor of HIV-1 reactivation. Moreover, in CD4+ T cells from aviremic HIV-1-positive donors under cART, HIV-1 proviral reactivation is unaffected by the addition of rapamycin, even if the drug downregulates markers of toxicity related to inflammation [96]. At the same time, active mTORC1 might be important for natural HIV-1 suppression control, because mTORC1 activation was also detected in HIV-1 elite controllers, a rare (<1%) group of HIV-1-infected patients who do not take antiretroviral therapy (ART) and do not develop AIDS [97].
The use of rapamycin and pan-inhibitors of mTORC1 is associated with multiple adverse effects; therefore, targeting the mTORC1 signaling pathway and not mTOR kinase itself appears to be an attractive alternative approach. Recent genome-wide CRISPR screening for the host factors required for HIV-1 latency identified two upstream mTORC1 inhibitory genes: TSC1 and DEPDC5; both are known negative regulators of mTORC1, but they suppress mTORC1 via different upstream branches. TSC1, a TSC complex member, inhibits mTORC1 and maintains HIV-1 latency by downregulation of RHEB, whereas DEPDC5, GATOR1 component, does so via suppression of RagA [98]. Therefore, targeting of TSC1 and DEPDC5 might be a useful strategy in metabolic inhibition to “block and lock” the latent reservoir.
In an effort to optimize the “shock and kill” approach, recent findings revealed that interaction of infected CD4+ T cells with DC could activate latent HIV-1 [99]. The contact with dendritic cells also activates the PI3K-Akt-mTOR pathway in CD4+ T cells and contributes to HIV-1 purge.
CD4+ T cells express on their surface various receptors that target them to different peripheral tissues in the body. One such receptor, CCR6, directs Th17 T-helper cells to the gut. The gut-associated lymphoid tissues are considered an important site of HIV-1 replication and a viral reservoir. The analysis of colon biopsies taken from HIV-1 patients under cART revealed that CCR6+ Th17-polarized CD4+ T cells express more mTOR, and that mTOR phosphorylation is also increased in these cells [53]. The use of mTOR inhibitors limited HIV-1 replication in gut-homing Th17 cells during reverse transcription and prior integration, with mTORC1/mTORC2 inhibitor INK128 being more effective than rapamycin. Thus, mTOR inhibitors could have a potential beneficial effect in decreasing HIV-1 reservoirs and restoring Th17 immunity in intestines during cART.
Finally, recent mathematical models predict that the persistence of a majority of HIV-1 infected cells is due to cellular proliferation rather than HIV-1 replication [100]; therefore, reducing cell proliferation could decrease the size of the HIV-1 reservoir. mTORC1 is a major regulator of cellular proliferation, and the specific targeting of this pathway in HIV-1 latent cells appears to be a promising approach by which to cure AIDS.
7. mTORC1 Pathway in HIV-1-Related Diseases
HIV-1-infected individuals have an elevated risk of developing of so-called AIDS-defining cancers: Kaposi’s sarcoma, B-cell lymphomas and cervical cancer [101] (Figure 3). Current antiretroviral therapy efficiently suppresses HIV-1, but high incidence of HIV-1-associated malignancies persists, although this has sharply declined in developed countries since the introduction of cART [86]. Thus, it is important to understand the mechanisms of the development of HIV-1-related diseases, even in patients under cART.
Kaposi’s sarcoma is caused by a herpesvirus (KSHV or HHV8). Ectopic expression of the viral proteins K1 and vGPCR in primary human umbilical vein endothelial cells (HUVEC) was shown to activate PI3K-AKT-mTOR pathway, which plays a central role in Kaposi’s sarcomagenesis [51,102,103,104]. KSHV infection selectively upregulates mTORC1 signaling in primary human endothelial cells (LECs), but not in blood endothelial cells [105]. KSHV infection of LECs makes these cells dependent on the mTORC1 pathway for their survival, because the treatment of these cells with rapamycin triggers apoptosis. Interestingly, primary effusion lymphoma, a tumor of B cell origin linked to KSHV infection, was strongly inhibited by rapamycin both in vitro and in mouse models [106].
Although HIV-1 does not infect B lymphocytes, the most frequent AIDS-related lymphomas (ARLs) are always of B cell origin [107]. One of the reasons for these malignancies might the genomic instability and chromatin remodeling in B cells caused by the entire HIV-1 and HIV-1 Tat protein, which is present in the blood of HIV-1-infected individuals and can penetrate different cells, including B cells [59,108]. Another possibility might involve the mTORC1 pathway, because studies in cell lines, primary cultures and tissue samples from patients have shown that mTORC1 is hyperactivated in diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma (MCL), Hodgkin lymphoma (HL), Burkitt lymphoma (BL) and anaplastic large cell lymphoma [109,110,111,112,113,114]. In order to inhibit mTORC1, rapalogues were used in clinical trials in MCL, DLBCL, HL and indolent lymphoma, but failed to provide substantial benefits for patients [115,116,117,118,119]. This failure is not specific to B-cell malignancies, but also concerns many other cancers. The main reason for rapalogue inefficiency is related to the activation of alternative proliferation pathways and genetic and functional intratumoral heterogeneity of mTORC1 activity [120]. Recent advances in the development of alternative mTOR inhibitors (pan-inhibitors and dual inhibitors) and the results of combination therapies with DNA damaging anticancer drugs should bring an essential breakthrough in the treatment of these cancers [8]. Indeed, the simultaneous inhibition of Akt with Nelfinavir and MK-2206 and mTORC1 with rapamycin showed a synergistic effect in suppressing DLBCL [113,121].
Similarly, the combination of mTORC1 inhibitors with different anti-HIV-1 drugs showed significant effects in suppressing viral replication, even rendering the antiviral drug-resistant strains sensitive to medication in T cells, PBMCs and in a humanized mouse model [15,48,122,123].
One of the frequent complications occurring typically in young adults of African ancestry with advanced HIV-1 is a HIV-associated nephropathy (HIVAN). The role of mTORC1 in HIVAN was addressed both in HIVAN murine models and upon analysis of tissue samples obtained from renal cortical sections of HIVAN patients [124,125,126,127,128]. Both HIVAN mice and HIVAN patients exhibit the activation of the mTORC1 pathway, as shown by elevated phosphorylation of mTOR itself and its substrates p70SK1 and 4EBP1. Treatment with rapamycin not only attenuated the HIVAN phenotype, but also partially inhibited tubular cell protein synthesis, specific miRNA expression and renal lesions [124,125,126]. In addition, inhibition of the mTORC1 pathway in HIVAN mice resulted in the downregulation of renal tissue p53 expression, and provided protection against p53-mediated oxidative kidney cell injury [127].
Besides the suppression of HIV-1 replication, cART drugs have several additional pharmacological activities that interfere with the mTORC1 pathway. A canonical cART is a combination of two nucleoside reverse transcriptase inhibitors (NRTIs) with one of the following drugs: HIV-protease inhibitors (PIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs) or HIV-integrase strand transfer inhibitors (INSTIs) [129]. Other FDA-approved classes of antiretroviral drugs are fusion inhibitors, CCR5 antagonists, CD4 post-attachment inhibitors [129]. PIs act via different mechanisms, mainly associated with the inhibition of cellular proteasome machinery (Hsp90 inhibition, ER stress). This leads to decreased PI3K-AKT signaling and downstream mTORC1 downregulation [130,131,132] (Figure 3). CCR5 antagonists also disrupt the PI3K-AKT-mTORC1 pathway [133,134]. In contrast, the side action of NRTIs can implicate active mTORC1 signaling in the host cell, although a direct link between them has not yet been established. The administration of NRTIs may cause severe mitochondrial toxicity [135], related to their moderate affinity to host DNA polymerases, especially the mitochondrial polymerase γ [136]. Mitochondrial toxicity can lead to the inhibition of late stages of autophagy mediated by active mTORC1 signaling, as shown for zidovudine (NRTI) in myocytes [137]. Another study demonstrated that stavudine (NRTI) could activate mTORC1 signaling in a mouse model of peripheral neuropathic pain [138]. Treatment with rapamycin attenuates persistent neuropathic pain in mice. Additionally, several cART drugs, being substrates of the CYP450 enzyme family, can modify the metabolism of mTORC1 inhibitors when used concomitantly, leading to altered mTORC1 axis regulation in patients [139].
8. Concluding Remarks
As a central integrator of various stimuli from intra- and extra- cellular environments, mTORC1 plays a vital role both in physiological and pathological conditions, including viral infections and cancers. In the immune system, mTORC1 senses cues from the immune microenvironment and influences the differentiation and maturation of immune cells. During HIV-1 infection, the mTORC1 pathway can be modulated not only by the entire virus, but also by its proteins. A combination of anti-HIV drugs with inhibitors of the mTORC1 pathway might be an efficient measure to cure, rather than control, HIV-1 replication.
9. Outstanding Questions
What are the molecular mechanisms of the interaction of HIV-1 and its separate proteins with the components of the mTORC1 pathway?
Are there ways to efficiently and specifically control mTORC1 suppression (and autophagy activation) in host cells?
Will a new generation of mTORC1 inhibitors provide additional benefits in overcoming of HIV-1 latency?
To what extent is the mTORC1 pathway involved in the appearance and persistence of HIV-related malignances in patients under cART?
Funding
This research was funded by French agencies INSERM (Enviburkitt) and ANRS. B.A. was funded by PhD exchange program between Kazakhstan and France.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eisenreich, W.; Rudel, T.; Heesemann, J.; Goebel, W. How Viral and Intracellular Bacterial Pathogens Reprogram the Metabolism of Host Cells to Allow Their Intracellular Replication. Front. Cell. Infect. Microbiol. 2019, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef]
- Votteler, J.; Schubert, U. Human Immunodeficiency Viruses: Molecular Biology. In Encyclopedia of Virology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 517–525. [Google Scholar]
- Burdick, R.C.; Li, C.; Munshi, M.H.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Ma, Y.; Vassetzky, Y.; Dokudovskaya, S. mTORC1 pathway in DNA damage response. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 1293–1311. [Google Scholar] [CrossRef]
- Cervera, C.; Cofan, F.; Hernandez, C.; Soy, D.; Marcos, M.A.; Sanclemente, G.; Bodro, M.; Moreno, A.; Diekmann, F.; Campistol, J.M.; et al. Effect of mammalian target of rapamycin inhibitors on cytomegalovirus infection in kidney transplant recipients receiving polyclonal antilymphocyte globulins: A propensity score-matching analysis. Transpl. Int. 2016, 29, 1216–1225. [Google Scholar] [CrossRef]
- Piñana, J.L.; Perez-Pitarch, A.; Guglieri-Lopez, B.; Giménez, E.; Hernandez-Boluda, J.C.; Terol, M.J.; Ferriols-Lisart, R.; Solano, C.; Navarro, D. Sirolimus exposure and the occurrence of cytomegalovirus DNAemia after allogeneic hematopoietic stem cell transplantation. Am. J. Transplant. 2018, 18, 2885–2894. [Google Scholar] [CrossRef] [Green Version]
- Mannick, J.B.; Morris, M.; Hockey, H.-U.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef] [Green Version]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV -chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donia, M.; McCubrey, J.A.; Bendtzen, K.; Nicoletti, F. Potential use of rapamycin in HIV infection. Br. J. Clin. Pharmacol. 2010, 70, 784–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, F.; Fagone, P.; Meroni, P.; McCubrey, J.; Bendtzen, K. mTOR as a multifunctional therapeutic target in HIV infection. Drug Discov. Today 2011, 16, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Vander Haar, E.; Lee, S.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Dokudovskaya, S.; Rout, M.P. SEA you later alli-GATOR--a dynamic regulator of the TORC1 stress response pathway. J. Cell Sci. 2015, 128, 2219–2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2016, 129, 1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaner, S.; Sodhi, A.; Pece, S.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001, 61, 2641–2648. [Google Scholar]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of Immune Responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.N.; Ramírez-Komo, J.A.; Park, H.; Iritani, B.M. Title: Control of B Lymphocyte Development and Functions by the mTOR Signaling Pathways. Cytokine Growth Factor Rev. 2017, 35, 47–62. [Google Scholar] [CrossRef]
- Liu, C.; Chapman, N.M.; Karmaus, P.W.F.; Zeng, H.; Chi, H. mTOR and metabolic regulation of conventional and regulatory T cells. J. Leukoc. Biol. 2015, 97, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurebayashi, Y.; Nagai, S.; Ikejiri, A.; Ohtani, M.; Ichiyama, K.; Baba, Y.; Yamada, T.; Egami, S.; Hoshii, T.; Hirao, A.; et al. PI3K-Akt-mTORC1-S6K1/2 Axis Controls Th17 Differentiation by Regulating Gfi1 Expression and Nuclear Translocation of RORγ. Cell Rep. 2012, 1, 360–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Shrestha, S.; Zeng, H.; Karmaus, P.W.F.; Neale, G.; Vogel, P.; Guertin, D.A.; Lamb, R.F.; Chi, H. T Cell Exit from Quiescence and Differentiation into Th2 Cells Depend on Raptor-mTORC1-Mediated Metabolic Reprogramming. Immunity 2013, 39, 1043–1056. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.; Jin, H.-S.; Lopez, J.; Elly, C.; Kim, G.; Murai, M.; Kronenberg, M.; Liu, Y.-C. TSC1 regulates the balance between effector and regulatory T cells. J. Clin. Investig. 2013, 123, 5165–5178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.-B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.-H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.-C. Inflammatory T Cell Responses Rely on Amino Acid Transporter ASCT2 Facilitation of Glutamine Uptake and mTORC1 Kinase Activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [Green Version]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR Kinase Determines Effector versus Memory CD8 + T Cell Fate by Regulating the Expression of Transcription Factors T-bet and Eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Yang, K.; Cloer, C.; Neale, G.; Vogel, P.; Chi, H. mTORC1 couples immune signals and metabolic programming to establish Treg-cell function. Nature 2013, 499, 485–490. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Warmoes, M.O.; de Cubas, A.A.; MacIver, N.J.; Locasale, J.W.; et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, T.N.; Ramírez, J.A.; Tsang, M.; Park, H.; Margineantu, D.H.; Hockenbery, D.M.; Iritani, B.M. Conditional Disruption of Raptor Reveals an Essential Role for mTORC1 in B Cell Development, Survival, and Metabolism. J. Immunol. 2016, 197, 2250–2260. [Google Scholar] [CrossRef] [Green Version]
- Raybuck, A.L.; Cho, S.H.; Li, J.; Rogers, M.C.; Lee, K.; Williams, C.L.; Shlomchik, M.; Thomas, J.W.; Chen, J.; Williams, J.V.; et al. B Cell–Intrinsic mTORC1 Promotes Germinal Center–Defining Transcription Factor Gene Expression, Somatic Hypermutation, and Memory B Cell Generation in Humoral Immunity. J. Immunol. 2018, 200, 2627–2639. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Pruitt, M.; Tran, D.; Du Bois, W.; Zhang, K.; Patel, R.; Hoover, S.; Simpson, R.M.; Simmons, J.; Gary, J.; et al. B Cell–Specific Deficiencies in mTOR Limit Humoral Immune Responses. J. Immunol. 2013, 191, 1692–1703. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.S.; Zhang, Z.-G.; Zhou, Z.; Du, G.-Y.; Li, H.; Yu, X.-Y.; Huang, Y.-H. PKM2-mediated inhibition of autophagy facilitates Tat’s inducing HIV-1 transactivation. Arch. Biochem. Biophys. 2017, 625–626, 17–23. [Google Scholar] [CrossRef]
- Kumar, B.; Arora, S.; Ahmed, S.; Banerjea, A.C. Hyperactivation of mammalian target of rapamycin complex 1 by HIV-1 is necessary for virion production and latent viral reactivation. FASEB J. 2017, 31, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 inhibits autophagy in bystander macrophage/ monocytic cells through Src-Akt and STAT3. PLoS ONE 2010, 5, e11733. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Yao, S.; Hu, M.; Li, W.; Hao, T.; Zhou, F.; Zhu, X.; Lu, H.; Qin, D.; Yan, Q.; et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014, 42, 9862–9879. [Google Scholar] [CrossRef]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Valiente-Echeverría, F.; Crossie, C.; Miron, M.-J.; Panté, N.; Olivier, M.; Mouland, A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Sci. Rep. 2017, 7, 5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Planas, D.; Routy, J.; Ancuta, P.; Planas, D.; Zhang, Y.; Monteiro, P.; Goulet, J.; Gosselin, A. HIV-1 selectively targets gut-homing mechanisms Find the latest version: HIV-1 selectively targets gut-homing mechanisms. JCI Insight 2017, 2, e93230. [Google Scholar] [CrossRef] [PubMed]
- Molle, D.; Segura-Morales, C.; Camus, G.; Berlioz-Torrent, C.; Kjems, J.; Basyuk, E.; Bertrand, E. Endosomal trafficking of HIV-1 gag and genomic RNAs regulates viral egress. J. Biol. Chem. 2009, 284, 19727–19743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvez, M.; Hseeh, G.; Benzer, S.; Brown, A.M. Osteopontin counters human immunodeficiency virus type 1–induced impairment of neurite growth through mammalian target of rapamycin and beta-integrin signaling pathways. J. Neurovirol. 2019, 25, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, W.; Tong, P.; Leung, C.-K.; Yang, G.; Li, Z.; Li, N.; Sun, X.; Han, Y.; Lu, C.; et al. Autophagy Induction by HIV-Tat and Methamphetamine in Primary Midbrain Neuronal Cells of Tree Shrews via the mTOR Signaling and ATG5/ATG7 Pathway. Front. Neurosci. 2018, 12, 921. [Google Scholar] [CrossRef]
- Poggi, A.; Carosio, R.; Fenoglio, D.; Brenci, S.; Murdaca, G.; Setti, M.; Indiveri, F.; Scabini, S.; Ferrero, E.; Zocchi, M.R. Migration of V delta 1 and V delta 2 T cells in response to CXCR3 and CXCR4 ligands in healthy donors and HIV-1-infected patients: Competition by HIV-1 Tat. Blood 2004, 103, 2205–2213. [Google Scholar] [CrossRef]
- Germini, D.; Tsfasman, T.; Klibi, M.; El-Amine, R.; Pichugin, A.; Iarovaia, O.V.; Bilhou-Nabera, C.; Subra, F.; Bou Saada, Y.; Sukhanova, A.; et al. HIV Tat induces a prolonged MYC relocalization next to IGH in circulating B-cells. Leukemia 2017, 31, 2515–2522. [Google Scholar] [CrossRef]
- Kurnaeva, M.A.; Sheval, E.V.; Musinova, Y.R.; Vassetzky, Y.S. Tat basic domain: A “Swiss army knife” of HIV-1 Tat? Rev. Med. Virol. 2019, 29, e2031. [Google Scholar] [CrossRef]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef] [Green Version]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel) 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Beek, N.; Klionsky, D.J.; Reggiori, F. Genetic aberrations in macroautophagy genes leading to diseases. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of autophagy in HIV infection and pathogenesis. J. Intern. Med. 2017, 281, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, É.; Barbeau, B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef] [Green Version]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy 2020, 8627, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Young, L.N.; Morris, K.L.; von Bülow, S.; Schöneberg, J.; Yamamoto-Imoto, H.; Oe, Y.; Yamamoto, K.; Nakamura, S.; Stjepanovic, G.; et al. Bidirectional Control of Autophagy by BECN1 BARA Domain Dynamics. Mol. Cell 2019, 73, 339–353. [Google Scholar] [CrossRef] [Green Version]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- Espert, L.; Varbanov, M.; Robert-Hebmann, V.; Sagnier, S.; Robbins, I.; Sanchez, F.; Lafont, V.; Biard-Piechaczyk, M. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 2009, 4, e5787. [Google Scholar] [CrossRef]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy Restricts HIV-1 Infection by Selectively Degrading Tat in CD4 + T Lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Herns, S.D.; Joshi, S.; Durden, D.L.; Spector, S.A. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. J. Biol. Chem. 2018, 293, 5808–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaisal, J.; Machado, A.; Galais, M.; Robert-Hebmann, V.; Arnauné-Pelloquin, L.; Espert, L.; Biard-Piechaczyk, M. HIV-1 Vpr inhibits autophagy during the early steps of infection of CD4 T cells. Biol. Cell 2019, 111, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Borel, S.; Robert-Hebmann, V.; Alfaisal, J.; Jain, A.; Faure, M.; Espert, L.; Chaloin, L.; Paillart, J.-C.; Johansen, T.; Biard-Piechaczyk, M. HIV-1 viral infectivity factor interacts with microtubule-associated protein light chain 3 and inhibits autophagy. AIDS 2015, 29, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Yakasai, A.M. Impact of Symptomatic HIV- Related Neurocognitive Disorders in Survival of HIV- Infected Individuals: A Systematic Review and Meta-Analyses. J. Neuroinfect. Dis. 2015, 06, 1–7. [Google Scholar] [CrossRef]
- Fields, J.; Dumaop, W.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat Alters Neuronal Autophagy by Modulating Autophagosome Fusion to the Lysosome: Implications for HIV-Associated Neurocognitive Disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.P.; De Simone, F.I.; Iorio, V.; De Marco, M.; Khalili, K.; Sariyer, I.K.; Capunzo, M.; Nori, S.L.; Rosati, A. HIV-1 Tat protein induces glial cell autophagy through enhancement of BAG3 protein levels. Cell Cycle 2014, 13, 3640–3644. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Dong, H.; Ye, X.; Zhong, L.; Cao, T.; Xu, Q.; Wang, J.; Zhang, Y.; Xu, J.; Wang, W.; et al. HIV-1 Tat increases BAG3 via NF-κB signaling to induce autophagy during HIV-associated neurocognitive disorder. Cell Cycle 2018, 17, 1614–1623. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.F.; Li, Q.; Li, J.; Wong, N.; Li, Z.; Huang, J.; Yang, G.; Sham, P.C.; Li, S.B.; Lu, G. HIV-1 Tat and methamphetamine co-induced oxidative cellular injury is mitigated by N-acetylcysteine amide (NACA) through rectifying mTOR signaling. Toxicol. Lett. 2018, 299, 159–171. [Google Scholar] [CrossRef]
- Qi, L.; Gang, L.; Hang, K.W.; Ling, C.H.; Xiaofeng, Z.; Zhen, L.; David Wai, Y.; Sang, P.W. Programmed neuronal cell death induced by HIV-1 tat and methamphetamine. Microsc. Res. Tech. 2011, 74, 1139–1144. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sereti, I.; Krebs, S.J.; Phanuphak, N.; Fletcher, J.L.; Slike, B.; Pinyakorn, S.; O’Connell, R.J.; Rupert, A.; Chomont, N.; Valcour, V.; et al. Persistent, Albeit Reduced, Chronic Inflammation in Persons Starting Antiretroviral Therapy in Acute HIV Infection. Clin. Infect. Dis. 2017, 64, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Guardigni, V.; Pencina, K.M.; Amato, A.A.; Floyd, M.; Brawley, B.; Mozeleski, B.; McKinnon, J.; Woodbury, E.; Heckel, E.; et al. Atypical Skeletal Muscle Profiles in Human Immunodeficiency Virus-Infected Asymptomatic Middle-Aged Adults. Clin. Infect. Dis. 2018, 66, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Thrift, A.P.; Chiao, E.Y. Are Non-HIV Malignancies Increased in the HIV-Infected Population? Curr. Infect. Dis. Rep. 2018, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Shmakova, A.; Germini, D.; Vassetzky, Y. HIV-1, HAART and cancer: A complex relationship. Int. J. Cancer 2020, 146, 2666–2679. [Google Scholar] [CrossRef] [PubMed]
- Hamers, R.L.; Rinke de Wit, T.F.; Holmes, C.B. HIV drug resistance in low-income and middle-income countries. Lancet HIV 2018, 5, e588–e596. [Google Scholar] [CrossRef]
- Chimukangara, B.; Lessells, R.J.; Rhee, S.Y.; Giandhari, J.; Kharsany, A.B.M.; Naidoo, K.; Lewis, L.; Cawood, C.; Khanyile, D.; Ayalew, K.A.; et al. Trends in Pretreatment HIV-1 Drug Resistance in Antiretroviral Therapy-naive Adults in South Africa, 2000–2016: A Pooled Sequence Analysis. EClinicalMedicine 2019, 9, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The Challenge of Viral Reservoirs in HIV-1 Infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef]
- Eisele, E.; Siliciano, R.F. Redefining the Viral Reservoirs that Prevent HIV-1 Eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, M.M.; Tang, Y.; Li, D.; Jiang, G. Deep latency: A new insight into a functional HIV cure. EBioMedicine 2019, 45, 624–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Investig. 2017, 127, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, F.Z.; Ouyang, Z.; Buzon, M.; Walker, B.D.; Lichterfeld, M.; Yu, X.G. Metabolic pathway activation distinguishes transcriptional signatures of CD8+ T cells from HIV-1 elite controllers. AIDS 2018, 32, 2669–2677. [Google Scholar] [CrossRef]
- Jin, S.; Liao, Q.; Chen, J.; Zhang, L.; He, Q.; Zhu, H.; Zhang, X.; Xu, J. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg. Microbes Infect. 2018, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Van Montfort, T.; van der Sluis, R.; Darcis, G.; Beaty, D.; Groen, K.; Pasternak, A.O.; Pollakis, G.; Vink, M.; Westerhout, E.M.; Hamdi, M.; et al. Dendritic cells potently purge latent HIV-1 beyond TCR-stimulation, activating the PI3K-Akt-mTOR pathway. EBioMedicine 2019, 42, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Reeves, D.B.; Duke, E.R.; Wagner, T.A.; Palmer, S.E.; Spivak, A.M.; Schiffer, J.T. A majority of HIV persistence during antiretroviral therapy is due to infected cell proliferation. Nat. Commun. 2018, 9, 4811. [Google Scholar] [CrossRef] [Green Version]
- Shiels, M.S.; Engels, E.A. Evolving epidemiology of HIV-associated malignancies. Curr. Opin. HIV AIDS 2017, 12, 6–11. [Google Scholar] [CrossRef]
- Tomlinson, C.C.; Damania, B. The K1 Protein of Kaposi’s Sarcoma-Associated Herpesvirus Activates the Akt Signaling Pathway. J. Virol. 2004, 78, 1918–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaner, S. Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: Emerging insights into the molecular oncogenesis and treatment of Kaposi’s sarcoma. Cell Cycle 2007, 6, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, D.A.; Busarla, S.V.P.; Gimbel, D.C.; Sohani, A.R.; Nazarian, R.M. mTOR, VEGF, PDGFR, and c-kit signaling pathway activation in Kaposi sarcoma. Hum. Pathol. 2017, 65, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Ganem, D. A Unique Herpesviral Transcriptional Program in KSHV-Infected Lymphatic Endothelial Cells Leads to mTORC1 Activation and Rapamycin Sensitivity. Cell Host Microbe 2013, 13, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, S.-H.; Roy, D.; Wang, L.; Staudt, M.R.; Fakhari, F.D.; Patel, D.D.; Henry, D.; Harrington, W.J.; Damania, B.A.; Dittmer, D.P. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 2007, 109, 2165–2173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Re, A.; Cattaneo, C.; Rossi, G. HIV and lymphoma: From epidemiology to clinical management. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019004. [Google Scholar] [CrossRef]
- Wang, X.; Duan, Z.; Yu, G.; Fan, M.; Scharff, M.D. Human Immunodeficiency Virus Tat Protein Aids V Region Somatic Hypermutation in Human B Cells. MBio 2018, 9, e02315–e02317. [Google Scholar] [CrossRef] [Green Version]
- El-Salem, M.; Raghunath, P.N.; Marzec, M.; Liu, X.; Kasprzycka, M.; Robertson, E.; Wasik, M.A. Activation of mTORC1 signaling pathway in AIDS-related lymphomas. Am. J. Pathol. 2009, 175, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Sebestyén, A.; Sticz, T.B.; Márk, Á.; Hajdu, M.; Timár, B.; Nemes, K.; Nagy, N.; Váradi, Z.; Kopper, L. Activity and complexes of mTOR in diffuse large B-cell lymphomas—A tissue microarray study. Mod. Pathol. 2012, 25, 1623–1628. [Google Scholar] [CrossRef] [Green Version]
- Ágnes Márk, M.H.; Zsófia, V.; Sticz, T.B.; Nagy, N.; Csomor, J.; Berczi, L.; Varga, V.; Csóka, M.; Sebestyén, L.K.A. Characteristic mTOR activity in Hodgkin-lymphomas offers a potential therapeutic target in high risk disease—A combined tissue microarray, in vitro and in vivo study. BMC Cancer 2013, 13, 250. [Google Scholar]
- Browne, S.H.; Diaz-Perez, J.A.; Preziosi, M.; King, C.C.; Jones, G.A.; Jain, S.; Sun, X.; Reid, E.G.; Vandenberg, S.; Wang, H.Y. mTOR activity in AIDS-related diffuse large B-cell lymphoma. PLoS ONE 2017, 12, e0170771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Col, J.D.; Zancai, P.; Terrin, L.; Guidoboni, M.; Ponzoni, M.; Pavan, A.; Spina, M.; Bergamin, S.; Rizzo, S.; Tirelli, U.; et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood 2008, 111, 5142–5151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekihara, K.; Saitoh, K.; Han, L.; Ciurea, S.; Yamamoto, S.; Kikkawa, M.; Kazuno, S.; Taka, H.; Kaga, N.; Arai, H.; et al. Targeting mantle cell lymphoma metabolism and survival through simultaneous blockade of mTOR and nuclear transporter exportin-1. Oncotarget 2017, 8, 34552–34564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, G.; Herbrecht, R.; Romaguera, J.; Verhoef, G.; Crump, M.; Gisselbrecht, C.; Laurell, A.; Offner, F.; Strahs, A.; Berkenblit, A.; et al. Phase III Study to Evaluate Temsirolimus Compared With Investigator’s Choice Therapy for the Treatment of Relapsed or Refractory Mantle Cell Lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Reeder, C.B.; LaPlant, B.R.; Gupta, M.; Johnston, P.B.; Micallef, I.N.; Porrata, L.F.; Ansell, S.M.; Colgan, J.P.; Jacobsen, E.D.; et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia 2011, 25, 341–347. [Google Scholar] [CrossRef]
- Johnston, P.B.; LaPlant, B.; McPhail, E.; Habermann, T.M.; Inwards, D.J.; Micallef, I.N.; Colgan, J.P.; Nowakowski, G.S.; Ansell, S.M.; Witzig, T.E. Everolimus combined with R-CHOP-21 for new, untreated, diffuse large B-cell lymphoma (NCCTG 1085 [Alliance]): Safety and efficacy results of a phase 1 and feasibility trial. Lancet Haematol. 2016, 3, e309–e316. [Google Scholar] [CrossRef] [Green Version]
- Bennani, N.N.; LaPlant, B.R.; Ansell, S.M.; Habermann, T.M.; Inwards, D.J.; Micallef, I.N.; Johnston, P.B.; Porrata, L.F.; Colgan, J.P.; Markovic, S.N.; et al. Efficacy of the oral mTORC1 inhibitor everolimus in relapsed or refractory indolent lymphoma. Am. J. Hematol. 2017, 92, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Johnston, P.B.; Pinter-Brown, L.C.; Warsi, G.; White, K.; Ramchandren, R. Phase 2 study of everolimus for relapsed or refractory classical Hodgkin lymphoma. Exp. Hematol. Oncol. 2018, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 Inhibitors in Cancer Therapy: From Kinase Mutations to Intratumoral Heterogeneity of Kinase Activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef] [Green Version]
- Petrich, A.M.; Leshchenko, V.; Kuo, P.-Y.; Xia, B.; Thirukonda, V.K.; Ulahannan, N.; Gordon, S.; Fazzari, M.J.; Ye, B.H.; Sparano, J.A.; et al. Akt Inhibitors MK-2206 and Nelfinavir Overcome mTOR Inhibitor Resistance in Diffuse Large B-cell Lymphoma. Clin. Cancer Res. 2012, 18, 2534–2544. [Google Scholar] [CrossRef] [Green Version]
- Heredia, A.; Gilliam, B.; Latinovic, O.; Le, N.; Bamba, D.; DeVico, A.; Melikyan, G.B.; Gallo, R.C.; Redfield, R.R. Rapamycin reduces CCR5 density levels on CD4 T cells, and this effect results in potentiation of enfuvirtide (T-20) against R5 strains of human immunodeficiency virus type 1 in vitro. Antimicrob. Agents Chemother. 2007, 51, 2489–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heredia, A.; Latinovic, O.; Gallo, R.C.; Melikyan, G.; Reitz, M.; Le, N.; Redfield, R.R. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proc. Natl. Acad. Sci. USA 2008, 105, 20476–20481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Konkimalla, S.; Yadav, A.; Sataranatarajan, K.; Kasinath, B.S.; Chander, P.N.; Singhal, P.C. HIV-Associated Nephropathy. Am. J. Pathol. 2010, 177, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.; Husain, M.; Yadav, A.; Kasinath, B.S.; Malhotra, A.; Singhal, P.C. HIV-1 Promotes Renal Tubular Epithelial Cell Protein Synthesis: Role of mTOR Pathway. PLoS ONE 2012, 7, e30071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, K.; Rai, P.; Plagov, A.; Lan, X.; Mathieson, P.W.; Saleem, M.A.; Husain, M.; Malhotra, A.; Singhal, P.C. Rapamycin-induced modulation of miRNA expression is associated with amelioration of HIV-associated nephropathy (HIVAN). Exp. Cell Res. 2013, 319, 2073–2080. [Google Scholar] [CrossRef] [Green Version]
- Rai, P.; Plagov, A.; Lan, X.; Chandel, N.; Singh, T.; Lederman, R.; Ayasolla, K.R.; Mathieson, P.W.; Saleem, M.A.; Husain, M.; et al. mTOR plays a critical role in p53-induced oxidative kidney cell injury in HIVAN. Am. J. Physiol. Physiol. 2013, 305, F343–F354. [Google Scholar] [CrossRef] [Green Version]
- Rai, P.; Lederman, R.; Haque, S.; Rehman, S.; Kumar, V.; Sataranatrajan, K.; Malhotra, A.; Kasinath, B.S.; Singhal, P.C. Renin angiotensin system modulates mTOR pathway through AT2R in HIVAN. Exp. Mol. Pathol. 2014, 96, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Department of Health and Human Services. Panel on Antiretroviral Guidelines for Adults and Adolescents.Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Available online: https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf (accessed on 10 April 2020).
- Shim, J.S.; Rao, R.; Beebe, K.; Neckers, L.; Han, I.; Nahta, R.; Liu, J.O. Selective Inhibition of HER2-Positive Breast Cancer Cells by the HIV Protease Inhibitor Nelfinavir. JNCI J. Natl. Cancer Inst. 2012, 104, 1576–1590. [Google Scholar] [CrossRef] [Green Version]
- Srirangam, A.; Mitra, R.; Wang, M.; Gorski, J.C.; Badve, S.; Baldridge, L.; Hamilton, J.; Kishimoto, H.; Hawes, J.; Li, L.; et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin. Cancer Res. 2006, 12, 1883–1896. [Google Scholar] [CrossRef] [Green Version]
- Goda, J.; Pachpor, T.; Basu, T.; Chopra, S.; Gota, V. Targeting the AKT pathway: Repositioning HIV protease inhibitors as radiosensitizers. Indian J. Med. Res. 2016, 143, 145. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Casagrande, N. Inhibition of the CCL5/CCR5 axis against the progression of gastric cancer. Int. J. Mol. Sci. 2018, 19, 1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Rahbar, R.; Fish, E.N. CCL5 activation of CCR5 regulates cell metabolism to enhance proliferation of breast cancer cells. Open Biol. 2016, 6, 160122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.J. Off-target effects of drugs that disrupt human mitochondrial DNA maintenance. Front. Mol. Biosci. 2017, 4, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A Review of the Toxicity of HIV Medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Stankov, M.V.; Hegermann, J.; Budida, R.; Panayotova-Dimitrova, D.; Schmidt, R.E.; Behrensa, G.M.N. Zidovudine-mediated autophagy inhibition enhances mitochondrial toxicity in muscle cells. Antimicrob. Agents Chemother. 2019, 63, e01443-18. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Wu, L.Y. Influence of phosphatidylinositol-3-kinase/protein kinase b-mammalian target of rapamycin signaling pathway on the neuropathic pain complicated by nucleoside reverse transcriptase inhibitors for the treatment of HIV infection. Chin. Med. J. (Engl.) 2018, 131, 1849–1856. [Google Scholar] [CrossRef]
- Hardy, W.D. Fundamentals of HIV Medicine 2019; Oxford University Press: Oxford, UK, 2019; ISBN 9780190942519. [Google Scholar]
Figure 1.
Upstream and downstream mTORC1 signaling. mTORC1 can be stimulated by various cues. Amino acids activate mTORC1 through GATOR1/GATOR2 complexes and RAGs. Growth factors recruits PI3K to the plasma membrane, which results in subsequent AKT activation. AKT can stimulate mTORC1 either directly through the phosphorylation of its component PRAS40, or via phosphorylation and inhibition of TSC1 from the TSC complex, which leads to RHEB and mTORC1 activations. Under hypoxic condition, mTORC1 is repressed due to TSC activation via AMPK. TSC can also be stimulated (i.e., mTORC1 repressed) via p53 pathway during DNA damage. Viruses can activate mTORC1, especially at the late stage of infection, both upstream, via PI3K/AKT, or acting on downstream mTORC1 target 4E-BP1.
Figure 1.
Upstream and downstream mTORC1 signaling. mTORC1 can be stimulated by various cues. Amino acids activate mTORC1 through GATOR1/GATOR2 complexes and RAGs. Growth factors recruits PI3K to the plasma membrane, which results in subsequent AKT activation. AKT can stimulate mTORC1 either directly through the phosphorylation of its component PRAS40, or via phosphorylation and inhibition of TSC1 from the TSC complex, which leads to RHEB and mTORC1 activations. Under hypoxic condition, mTORC1 is repressed due to TSC activation via AMPK. TSC can also be stimulated (i.e., mTORC1 repressed) via p53 pathway during DNA damage. Viruses can activate mTORC1, especially at the late stage of infection, both upstream, via PI3K/AKT, or acting on downstream mTORC1 target 4E-BP1.

Figure 2.
Implication of various HIV-1 proteins in the modulation of autophagy in different cells.

Figure 3.
mTORC1 status in HIV-1-related diseases.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Akbay, B.; Shmakova, A.; Vassetzky, Y.; Dokudovskaya, S. Modulation of mTORC1 Signaling Pathway by HIV-1. Cells 2020, 9, 1090. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051090
AMA Style
Akbay B, Shmakova A, Vassetzky Y, Dokudovskaya S. Modulation of mTORC1 Signaling Pathway by HIV-1. Cells. 2020; 9(5):1090. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051090
Chicago/Turabian StyleAkbay, Burkitkan, Anna Shmakova, Yegor Vassetzky, and Svetlana Dokudovskaya. 2020. "Modulation of mTORC1 Signaling Pathway by HIV-1" Cells 9, no. 5: 1090. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051090
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.