Targeting the IGF-Axis for Cancer Therapy: Development and Validation of an IGF-Trap as a Potential Drug

Abstract
:1. Background Information
1.1. The Insulin-Like Growth Factor (IGF)-Axis
1.2. Hybrid Receptors and Crosstalk with Other Receptors
1.3. Targeting of the IGF-Axis for Cancer Therapy—The Rational
1.3.1. IGF-Targeting for Cancer Management: The Current Landscape and Overall Clinical Experience
Targeting the IGF-I Receptor
Targeting the IGF-Ligands
2. Traps in the Clinic—Advantages and Challenges
3. The IGF-Trap—A Stepwise Bioengineering Venture
3.1. IGF-IR Decoys-Background Information
3.2. The Incremental Production/Validation Process for an IGF-Trap
3.3. A 3rd Generation IGF-Trap—Properties, Bioactivity and Challenges
4. Targeting the IGF-IR in the Tumor Microenvironment
4.1. IGF-IR Is Expressed on Immune Cells and Plays a Role in Immunosuppression
4.2. Multiple Effects of the IGF-Trap on the Tumor Microenvironment
4.3. Future Prospective: The Case for Combinatorial Therapy with the IGF-Trap
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Butler, A.A.; Yakar, S.; Gewolb, I.H.; Karas, M.; Okubo, Y.; LeRoith, D. Insulin-like growth factor-I receptor signal transduction: At the interface between physiology and cell biology. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 121, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implications for drug design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, F.; Takahashi, S.-I. 40 years of IGF1: IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Choi, E.; Yu, H.; Bai, X.-c. Structural basis of the activation of type 1 insulin-like growth factor receptor. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocmé, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef]
- Warsito, D.; Sjöström, S.; Andersson, S.; Larsson, O.; Sehat, B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. Embo Rep. 2012, 13, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Aleksic, T.; Chitnis, M.M.; Perestenko, O.V.; Gao, S.; Thomas, P.H.; Turner, G.D.; Protheroe, A.S.; Howarth, M.; Macaulay, V.M. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res. 2010, 70, 6412–6419. [Google Scholar] [CrossRef] [Green Version]
- Codony-Servat, J.; Cuatrecasas, M.; Asensio, E.; Montironi, C.; Martínez-Cardús, A.; Marín-Aguilera, M.; Horndler, C.; Martínez-Balibrea, E.; Rubini, M.; Jares, P. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br. J. Cancer 2017, 117, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Clément, F.; Martin, A.; Venara, M.; de Luján Calcagno, M.; Mathó, C.; Maglio, S.; Lombardi, M.G.; Bergadá, I.; Pennisi, P.A. Type 1 IGF Receptor Localization in Paediatric Gliomas: Significant Association with WHO Grading and Clinical Outcome. Horm. Cancer 2018, 9, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Vella, V.; Milluzzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin Receptor Isoforms in Cancer. Int. J. Mol. Sci. 2018, 19, 3615. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Vigneri, R.; Costantino, A.; Frasca, F.; Ippolito, A.; Fujita-Yamaguchi, Y.; Siddle, K.; Goldfine, I.D.; Belfiore, A. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: Evidence for a second mechanism of IGF-I signaling. Clin. Cancer Res. 1999, 5, 1935–1944. [Google Scholar] [PubMed]
- Belfiore, A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 2007, 13, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Nagle, A.M.; Wang, Y.F.; Boone, D.N.; Lee, A.V. Controlled dimerization of insulin-like growth factor-1 and insulin receptors reveals shared and distinct activities of holo and hybrid receptors. J. Biol. Chem. 2018, 293, 3700–3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, D.M.; Hewitt, S.C.; Ciana, P.; Raviscioni, M.; Lindzey, J.K.; Foley, J.; Maggi, A.; DiAugustine, R.P.; Korach, K.S. Requirement of estrogen receptor-α in insulin-like growth factor-1 (IGF-1)-induced uterine responses and in vivo evidence for IGF-1/estrogen receptor cross-talk. J. Biol. Chem. 2002, 277, 8531–8537. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J.A.; Varewijck, A.J. IGF-IR targeted therapy: Past, present and future. Front. Endocrinol. 2014, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Sun, X.; Shen, B. Molecular imaging of IGF-1R in cancer. Mol. Imaging 2017, 16, 1536012117736648. [Google Scholar] [CrossRef] [Green Version]
- Shanmugalingam, T.; Bosco, C.; Ridley, A.J.; Van Hemelrijck, M. Is there a role for IGF-1 in the development of second primary cancers? Cancer Med. 2016, 5, 3353–3367. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Brodt, P.; Sun, H.; Mejia, W.; Novosyadlyy, R.; Nunez, N.; Chen, X.; Mendoza, A.; Hong, S.H.; Khanna, C.; et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 2010, 70, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakar, S.; Leroith, D.; Brodt, P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: Lessons from animal models. Cytokine Growth Factor Rev. 2005, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, Y.; Waters, M.J.; Brooks, A.J. Role of the growth hormone–IGF-1 axis in cancer. Expert Rev. Endocrinol. Metab. 2011, 6, 71–84. [Google Scholar] [CrossRef] [PubMed]
- De Ostrovich, K.K.; Lambertz, I.; Colby, J.K.; Tian, J.; Rundhaug, J.E.; Johnston, D.; Conti, C.J.; DiGiovanni, J.; Fuchs-Young, R. Paracrine overexpression of insulin-like growth factor-1 enhances mammary tumorigenesis in vivo. Am. J. Pathol. 2008, 173, 824–834. [Google Scholar] [CrossRef] [Green Version]
- DiGiovanni, J.; Kiguchi, K.; Frijhoff, A.; Wilker, E.; Bol, D.K.; Beltrán, L.; Moats, S.; Ramirez, A.; Jorcano, J.; Conti, C. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 3455–3460. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-Y.; Jin, Q.; Oh, S.-H.; Kim, E.S.; Yang, Y.J.; Lee, D.H.; Feng, L.; Behrens, C.; Prudkin, L.; Miller, Y.E. Elevated epithelial insulin-like growth factor expression is a risk factor for lung cancer development. Cancer Res. 2009, 69, 7439–7448. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Tang, H.; Liao, Y.; Xu, Q.; Liu, C.; Deng, Y.; Wang, J.; Wang, J.; Fu, X. Expression and clinical significance of insulin-like growth factor 1 in lung cancer tissues and perioperative circulation from patients with non-small-cell lung cancer. Curr. Oncol. 2016, 23, 12. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, C.; Casanova-Salas, I.; Calatrava, A.; García-Flores, M.; Garofalo, C.; Grilli, A.; Rubio-Briones, J.; Scotlandi, K.; López-Guerrero, J.A. Insulin-like growth factor 1 receptor affects the survival of primary prostate cancer patients depending on TMPRSS2-ERG status. BMC Cancer 2017, 17, 367. [Google Scholar] [CrossRef]
- Mu, L.; Tuck, D.; Katsaros, D.; Lu, L.; Schulz, V.; Perincheri, S.; Menato, G.; Scarampi, L.; Harris, L.; Yu, H. Favorable outcome associated with an IGF-1 ligand signature in breast cancer. Breast Cancer Res. Treat. 2012, 133, 321–331. [Google Scholar] [CrossRef]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef]
- Valentinis, B.; Baserga, R. IGF-I receptor signalling in transformation and differentiation. Mol. Pathol. 2001, 54, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Valentinis, B.; Romano, G.; Peruzzi, F.; Morrione, A.; Prisco, M.; Soddu, S.; Cristofanelli, B.; Sacchi, A.; Baserga, R. Growth and differentiation signals by the insulin-like growth factor 1 receptor in hemopoietic cells are mediated through different pathways. J. Biol. Chem. 1999, 274, 12423–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llanos, A.A.; Brasky, T.M.; Dumitrescu, R.G.; Marian, C.; Makambi, K.H.; Kallakury, B.V.; Spear, S.L.; Perry, D.J.; Convit, R.J.; Platek, M.E. Plasma IGF-1 and IGFBP-3 may be imprecise surrogates for breast concentrations: An analysis of healthy women. Breast Cancer Res. Treat. 2013, 138, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, N.J.; Slater, T.A.; Matthews, C.J.; Wheatcroft, S.B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 2019, 19, 86–96. [Google Scholar]
- Bohula, E.A.; Playford, M.P.; Macaulay, V.M. Targeting the type 1 insulin-like growth factor receptor as anti-cancer treatment. Anti-Cancer Drugs 2003, 14, 669–682. [Google Scholar] [CrossRef]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [Green Version]
- Osher, E.; Macaulay, V.M. Therapeutic targeting of the IGF axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-specific and tri-specific antibodies-the next big thing in solid tumor therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef]
- Schanzer, J.M.; Wartha, K.; Moessner, E.; Hosse, R.J.; Moser, S.; Croasdale, R.; Trochanowska, H.; Shao, C.; Wang, P.; Shi, L.; et al. XGFR*, a novel affinity-matured bispecific antibody targeting IGF-1R and EGFR with combined signaling inhibition and enhanced immune activation for the treatment of pancreatic cancer. mAbs 2016, 8, 811–827. [Google Scholar] [CrossRef] [Green Version]
- Quinn, D.I.; Baudin, E.; Demeure, M.J.; Fassnacht, M.; Hammer, G.D.; Poondru, S.; Fleege, T.; Rorig, R.; Berruti, A. International randomized, double-blind, placebo-controlled, phase 3 study of linsitinib (OSI-906, L) in patients (pts) with locally advanced or metastatic adrenocortical carcinoma (ACC). J. Clin. Oncol. 2014, 32, 4507. [Google Scholar] [CrossRef]
- Tian, D.; Kreeger, P.K. Analysis of the quantitative balance between insulin-like growth factor (IGF)-1 ligand, receptor, and binding protein levels to predict cell sensitivity and therapeutic efficacy. BMC Syst. Biol. 2014, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.; Reichert, J.M. Therapeutic Fc-fusion proteins and peptides as successful alternatives to antibodies. mAbs 2011, 3, 415–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, A.; Pollak, M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: Early clinical trial results and future directions. Oncogene 2009, 28, 3009–3021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avnet, S.; Sciacca, L.; Salerno, M.; Gancitano, G.; Cassarino, M.F.; Longhi, A.; Zakikhani, M.; Carboni, J.M.; Gottardis, M.; Giunti, A.; et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res. 2009, 69, 2443–2452. [Google Scholar] [CrossRef] [Green Version]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [Green Version]
- Haluska, P.; Worden, F.; Olmos, D.; Yin, D.; Schteingart, D.; Batzel, G.N.; Paccagnella, M.L.; de Bono, J.S.; Gualberto, A.; Hammer, G.D. Safety, tolerability, and pharmacokinetics of the anti-IGF-1R monoclonal antibody figitumumab in patients with refractory adrenocortical carcinoma. Cancer Chemother. Pharmacol. 2010, 65, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Spigel, D.R.; Chen, D.; Steins, M.B.; Engelman, J.A.; Schneider, C.P.; Novello, S.; Eberhardt, W.E.; Crino, L.; Habben, K.; et al. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4574–4580. [Google Scholar] [CrossRef] [Green Version]
- Cohen, B.D.; Baker, D.A.; Soderstrom, C.; Tkalcevic, G.; Rossi, A.M.; Miller, P.E.; Tengowski, M.W.; Wang, F.; Gualberto, A.; Beebe, J.S.; et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin. Cancer Res. 2005, 11, 2063–2073. [Google Scholar] [CrossRef] [Green Version]
- Frogne, T.; Jepsen, J.S.; Larsen, S.S.; Fog, C.K.; Brockdorff, B.L.; Lykkesfeldt, A.E. Antiestrogen-resistant human breast cancer cells require activated protein kinase B/Akt for growth. Endocr. -Relat. Cancer 2005, 12, 599–614. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.J.; Liang, S.J.; Guo, N.; Li, S.L.; Wu, A.M.; Giannini, S.; Sachdev, D.; Yee, D.; Brunner, N.; Ikle, D.; et al. Combined effects of tamoxifen and a chimeric humanized single chain antibody against the type I IGF receptor on breast tumor growth in vivo. Horm. Metab Res. 2003, 35, 836–842. [Google Scholar] [PubMed]
- Gee, J.M.; Robertson, J.F.; Gutteridge, E.; Ellis, I.O.; Pinder, S.E.; Rubini, M.; Nicholson, R.I. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr. Relat. Cancer 2005, 12 (Suppl. 1), S99–S111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.E.; Gee, J.M.; Taylor, K.M.; Barrow, D.; Williams, H.D.; Rubini, M.; Nicholson, R.I. Development of strategies for the use of anti-growth factor treatments. Endocr. -Relat. Cancer 2005, 12 (Suppl. 1), S173–S182. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Yuan, L.X.; Zhang, B.; Kobayashi, R.; Esteva, F.J. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005, 65, 11118–11128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camirand, A.; Zakikhani, M.; Young, F.; Pollak, M. Inhibition of insulin-like growth factor-1 receptor signaling enhances growth-inhibitory and proapoptotic effects of gefitinib (Iressa) in human breast cancer cells. Breast Cancer Res. BCR 2005, 7, R570–R579. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Loeffler, J.S.; Dyson, N.J. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res. 2002, 62, 200–207. [Google Scholar]
- Gualberto, A.; Pollak, M. Clinical development of inhibitors of the insulin-like growth factor receptor in oncology. Curr. Drug Targets 2009, 10, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Douglas, R.S.; Kahaly, G.J.; Patel, A.; Sile, S.; Thompson, E.H.Z.; Perdok, R.; Fleming, J.C.; Fowler, B.T.; Marcocci, C.; Marino, M.; et al. Teprotumumab for the Treatment of Active Thyroid Eye Disease. N. Engl. J. Med. 2020, 382, 341–352. [Google Scholar] [CrossRef]
- Markham, A. Teprotumumab: First Approval. Drugs 2020, 10, 1–4. [Google Scholar] [CrossRef]
- Ko, A.H.; Cubillo, A.; Kundranda, M.; Zafar, S.F.; Meiri, E.; Bendell, J.; Alguel, H.; Rivera Herrero, F.; Ahn, E.; Watkins, D.; et al. CARRIE: A randomized, double-blind, placebo-controlled phase II study of istiratumab (MM-141) plus nab-paclitaxel and gemcitabine versus nab-paclitaxel and gemcitabine in front-line metastatic pancreatic cancer. Ann. Oncol. 2018, 29 (Suppl. 8), viii720. [Google Scholar] [CrossRef]
- Kundranda, M.; Gracian, A.C.; Zafar, S.F.; Meiri, E.; Bendell, J.; Algul, H.; Rivera, F.; Ahn, E.R.; Watkins, D.; Pelzer, U.; et al. Randomized, double-blind, placebo-controlled phase II study of istiratumab (MM-141) plus nab-paclitaxel and gemcitabine versus nab-paclitaxel and gemcitabine in front-line metastatic pancreatic cancer (CARRIE). Ann. Oncol. 2020, 31, 79–87. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Lin, C.C.; Chen, L.T.; Corral, J.; Michalarea, V.; Rihawi, K.; Ong, M.; Lee, J.H.; Hsu, C.H.; Yang, J.C.; et al. Two first-in-human studies of xentuzumab, a humanised insulin-like growth factor (IGF)-neutralising antibody, in patients with advanced solid tumours. Br. J. Cancer 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, H.; Nishina, T.; Nogami, N.; Kozuki, T.; Yamagiwa, Y.; Yagawa, K. Phase I dose-escalation study evaluating safety, tolerability and pharmacokinetics of MEDI-573, a dual IGF-I/II neutralizing antibody, in Japanese patients with advanced solid tumours. Invest. New Drugs 2015, 33, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Rajah, R.; Valentinis, B.; Cohen, P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-β1 on programmed cell death through a p53-and IGF-independent mechanism. J. Biol. Chem. 1997, 272, 12181–12188. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Marzec, K.; Martin, J.; Baxter, R. The role of insulin-like growth factor binding protein-3 in the breast cancer cell response to DNA-damaging agents. Oncogene 2014, 33, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Jerome, L.; Alami, N.; Belanger, S.; Page, V.; Yu, Q.; Paterson, J.; Shiry, L.; Pegram, M.; Leyland-Jones, B. Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2–overexpressing breast tumors and potentiates Herceptin activity in vivo. Cancer Res. 2006, 66, 7245–7252. [Google Scholar] [CrossRef] [Green Version]
- Soh, C.L.; McNeil, K.; Owczarek, C.M.; Hardy, M.P.; Fabri, L.J.; Pearse, M.; Delaine, C.A.; Forbes, B.E. Exogenous administration of protease-resistant, non-matrix-binding IGFBP-2 inhibits tumour growth in a murine model of breast cancer. Br. J. Cancer 2014, 110, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [Green Version]
- Trieu, Y.; Wen, X.Y.; Skinnider, B.F.; Bray, M.R.; Li, Z.; Claudio, J.O.; Masih-Khan, E.; Zhu, Y.X.; Trudel, S.; McCart, J.A.; et al. Soluble interleukin-13Ralpha2 decoy receptor inhibits Hodgkin’s lymphoma growth in vitro and in vivo. Cancer Res. 2004, 64, 3271–3275. [Google Scholar] [CrossRef] [Green Version]
- Tseng, J.F.; Farnebo, F.A.; Kisker, O.; Becker, C.M.; Kuo, C.J.; Folkman, J.; Mulligan, R.C. Adenovirus-mediated delivery of a soluble form of the VEGF receptor Flk1 delays the growth of murine and human pancreatic adenocarcinoma in mice. Surgery 2002, 132, 857–865. [Google Scholar] [CrossRef]
- Messori, A.; Santarlasci, B.; Vaiani, M. New drugs for rheumatoid arthritis. N. Engl. J. Med. 2004, 351, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, H.M.; Throne, M.L.; Amar, N.J.; Sebai, M.; Kivitz, A.J.; Kavanaugh, A.; Weinstein, S.P.; Belomestnov, P.; Yancopoulos, G.D.; Stahl, N.; et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheum. 2008, 58, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, C.; Ferber, A.; Resnicoff, M.; Baserga, R. A soluble insulin-like growth factor I receptor that induces apoptosis of tumor cells in vivo and inhibits tumorigenesis. Cancer Res. 1996, 56, 4013–4020. [Google Scholar]
- Dubois, E.A.; Rissmann, R.; Cohen, A.F. Rilonacept and canakinumab. Br. J. Clin. Pharm. 2011, 71, 639–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechtner, V.; Karanikas, C.A.; García-Pérez, L.E.; Glaesner, W. A new approach to drug therapy: Fc-fusion technology. Prim Health Care 2017, 7. [Google Scholar]
- Kim, E.S.; Serur, A.; Huang, J.; Manley, C.A.; McCrudden, K.W.; Frischer, J.S.; Soffer, S.Z.; Ring, L.; New, T.; Zabski, S.; et al. Potent VEGF blockade causes regression of coopted vessels in a model of neuroblastoma. Proc. Natl. Acad. Sci. USA 2002, 99, 11399–11404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindzen, M.; Carvalho, S.; Starr, A.; Ben-Chetrit, N.; Pradeep, C.R.; Kostler, W.J.; Rabinkov, A.; Lavi, S.; Bacus, S.S.; Yarden, Y. A recombinant decoy comprising EGFR and ErbB-4 inhibits tumor growth and metastasis. Oncogene 2012, 31, 3505–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohl, W.R. Fusion Proteins for Half-Life Extension of Biologics as a Strategy to Make Biobetters. BioDrugs 2015, 29, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Marotte, H.; Cimaz, R. Etanercept - TNF receptor and IgG1 Fc fusion protein: Is it different from other TNF blockers? Expert Opin. Biol. Ther. 2014, 14, 569–572. [Google Scholar] [CrossRef] [Green Version]
- Ravetch, J.V.; Bolland, S. IgG Fc receptors. Annu. Rev. Immunol. 2001, 19, 275–290. [Google Scholar] [CrossRef]
- Czajkowsky, D.M.; Hu, J.; Shao, Z.; Pleass, R.J. Fc-fusion proteins: New developments and future perspectives. Embo Mol. Med. 2012, 4, 1015–1028. [Google Scholar] [CrossRef]
- Idusogie, E.E.; Wong, P.Y.; Presta, L.G.; Gazzano-Santoro, H.; Totpal, K.; Ultsch, M.; Mulkerrin, M.G. Engineered antibodies with increased activity to recruit complement. J. Immunol. 2001, 166, 2571–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimchi-Sarfaty, C.; Schiller, T.; Hamasaki-Katagiri, N.; Khan, M.A.; Yanover, C.; Sauna, Z.E. Building better drugs: Developing and regulating engineered therapeutic proteins. Trends Pharm. Sci 2013, 34, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Gao, X.; Gong, R. Engineering of Fc Fragments with Optimized Physicochemical Properties Implying Improvement of Clinical Potentials for Fc-Based Therapeutics. Front. Immunol. 2017, 8, 1860. [Google Scholar] [CrossRef] [PubMed]
- Dunn, S.E.; Ehrlich, M.; Sharp, N.J.; Reiss, K.; Solomon, G.; Hawkins, R.; Baserga, R.; Barrett, J.C. A dominant negative mutant of the insulin-like growth factor-I receptor inhibits the adhesion, invasion, and metastasis of breast cancer. Cancer Res. 1998, 58, 3353–3361. [Google Scholar]
- Sachdev, D.; Hartell, J.S.; Lee, A.V.; Zhang, X.; Yee, D. A dominant negative type I insulin-like growth factor receptor inhibits metastasis of human cancer cells. J. Biol. Chem. 2004, 279, 5017–5024. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.; Adachi, Y.; Yamamoto, H.; Imsumran, A.; Arimura, Y.; Endo, T.; Hinoda, Y.; Lee, C.T.; Nadaf, S.; Carbone, D.P.; et al. Insulin-like growth factor I receptor blockade enhances chemotherapy and radiation responses and inhibits tumour growth in human gastric cancer xenografts. Gut 2005, 54, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Samani, A.A.; Chevet, E.; Fallavollita, L.; Galipeau, J.; Brodt, P. Loss of tumorigenicity and metastatic potential in carcinoma cells expressing the extracellular domain of the type 1 insulin-like growth factor receptor. Cancer Res. 2004, 64, 3380–3385. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Fallavollita, L.; Nguyen, L.; Burnier, J.; Rafei, M.; Galipeau, J.; Yakar, S.; Brodt, P. Autologous bone marrow stromal cells genetically engineered to secrete an igf-I receptor decoy prevent the growth of liver metastases. Mol. Ther. 2009, 17, 1241–1249. [Google Scholar] [CrossRef]
- Wang, N.; Lu, Y.; Pinard, M.; Pilotte, A.; Gilbert, R.; Massie, B.; Brodt, P. Sustained production of a soluble IGF-I receptor by gutless adenovirus-transduced host cells protects from tumor growth in the liver. Cancer Gene 2013, 20, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Rayes, R.F.; Elahi, S.M.; Lu, Y.; Hancock, M.A.; Massie, B.; Rowe, G.E.; Aomari, H.; Hossain, S.; Durocher, Y.; et al. The IGF-Trap: Novel Inhibitor of Carcinoma Growth and Metastasis. Mol. Cancer Ther. 2015, 14, 982–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2018, 9, 15–32. [Google Scholar] [CrossRef] [PubMed]
- Strand, J.; Huang, C.T.; Xu, J. Characterization of Fc-fusion protein aggregates derived from extracellular domain disulfide bond rearrangements. J. Pharm Sci. 2013, 102, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Sadick, M.D.; Intintoli, A.; Quarmby, V.; McCoy, A.; Canova-Davis, E.; Ling, V. Kinase receptor activation (KIRA): A rapid and accurate alternative to end-point bioassays. J. Pharm. Biomed. Anal. 1999, 19, 883–891. [Google Scholar] [CrossRef]
- Chen, J.W.; Ledet, T.; Orskov, H.; Jessen, N.; Lund, S.; Whittaker, J.; De Meyts, P.; Larsen, M.B.; Christiansen, J.S.; Frystyk, J. A highly sensitive and specific assay for determination of IGF-I bioactivity in human serum. Am. J. Physiol. Endocrinol. Metab 2003, 284, E1149–E1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espelund, U.S.; Bjerre, M.; Hjortebjerg, R.; Rasmussen, T.R.; Lundby, A.; Hoeflich, A.; Folkersen, B.H.; Oxvig, C.; Frystyk, J. Insulin-Like Growth Factor Bioactivity, Stanniocalcin-2, Pregnancy-Associated Plasma Protein-A, and IGF-Binding Protein-4 in Pleural Fluid and Serum From Patients With Pulmonary Disease. J. Clin. Endocrinol Metab 2017, 102, 3526–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frystyk, J. Quantification of the GH/IGF-axis components: Lessons from human studies. Domest Anim. Endocrinol. 2012, 43, 186–197. [Google Scholar] [CrossRef]
- Vaniotis, G.; Moffett, S.; Sulea, T.; Wang, N.; Elahi, S.M.; Lessard, E.; Baardsnes, J.; Perrino, S.; Durocher, Y.; Frystyk, J.; et al. Enhanced anti-metastatic bioactivity of an IGF-TRAP re-engineered to improve physicochemical properties. Sci. Rep. 2018, 8, 17361. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.J. Insulin-like growth factor-I regulation of immune function: A potential therapeutic target in autoimmune diseases? Pharm. Rev. 2010, 62, 199–236. [Google Scholar] [CrossRef] [Green Version]
- Xuan, N.T.; Hoang, N.H.; Nhung, V.P.; Duong, N.T.; Ha, N.H.; Hai, N.V. Regulation of dendritic cell function by insulin/IGF-1/PI3K/Akt signaling through klotho expression. J. Recept. Signal. Transduct. 2016, 1–7. [Google Scholar] [CrossRef]
- Bilbao, D.; Luciani, L.; Johannesson, B.; Piszczek, A.; Rosenthal, N. Insulin-like growth factor-1 stimulates regulatory T cells and suppresses autoimmune disease. Embo Mol. Med. 2014, 6, 1423–1435. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, S.; Hugenschmidt, H.; Hagness, M.; Line, P.D.; Labori, K.J.; Wiedswang, G.; Tasken, K.; Aandahl, E.M. Regulatory T cells that co-express RORgammat and FOXP3 are pro-inflammatory and immunosuppressive and expand in human pancreatic cancer. Oncoimmunology 2016, 5, e1102828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, Y.; Sukhanov, S.; Shai, S.Y.; Danchuk, S.; Tang, R.; Snarski, P.; Li, Z.; Lobelle-Rich, P.; Wang, M.; Wang, D.; et al. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E-Deficient Mice. Circulation 2016, 133, 2263–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.P.; Minogue, A.M.; Falvey, A.; Lynch, M.A. Involvement of IGF-1 and Akt in M1/M2 activation state in bone marrow-derived macrophages. Exp. Cell Res. 2015, 335, 258–268. [Google Scholar] [CrossRef]
- Huang, C.T.; Chang, M.C.; Chen, Y.L.; Chen, T.C.; Chen, C.A.; Cheng, W.F. Insulin-like growth factors inhibit dendritic cell-mediated anti-tumor immunity through regulating ERK1/2 phosphorylation and p38 dephosphorylation. Cancer Lett 2015, 359, 117–126. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Flashner-Abramson, E.; Shalapour, S.; Zhong, Z.; Taniguchi, K.; Levitzki, A.; Karin, M. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene 2016, 35, 2634–2644. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Sarantopoulos, J.; Patnaik, A.; Papadopoulos, K.; Lin, C.C.; Rodon, J.; Murphy, B.; Roth, B.; McCaffery, I.; Gorski, K.S.; et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5800–5807. [Google Scholar] [CrossRef]
- Shaw, A.K.; Pickup, M.W.; Chytil, A.; Aakre, M.; Owens, P.; Moses, H.L.; Novitskiy, S.V. TGFbeta signaling in myeloid cells regulates mammary carcinoma cell invasion through fibroblast interactions. PLoS ONE 2015, 10, e0117908. [Google Scholar] [CrossRef]
- Fernandez, M.C.; Rayes, R.; Ham, B.; Wang, N.; Bourdeau, F.; Milette, S.; Lllemann, M.; Bird, N.; Majeed, A.; Xu, J.; et al. The type I insulin-like growth factor regulates the liver stromal response to metastatic colon carcinoma cells. Oncotarget 2017, 8, 52281–52293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayes, R.F.; Milette, S.; Fernandez, M.C.; Ham, B.; Wang, N.; Bourdeau, F.; Perrino, S.; Yakar, S.; Brodt, P. Loss of neutrophil polarization in colon carcinoma liver metastases of mice with an inducible, liver-specific IGF-I deficiency. Oncotarget 2018, 9, 15691–15704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnier, J.V.; Wang, N.; Michel, R.P.; Hassanain, M.; Li, S.; Lu, Y.; Metrakos, P.; Antecka, E.; Burnier, M.N.; Ponton, A.; et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene 2011, 30, 3766–3783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaniotis, G.; Rayes, R.F.; Qi, S.; Milette, S.; Wang, N.; Perrino, S.; Bourdeau, F.; Nystrom, H.; He, Y.; Lamarche-Vane, N.; et al. Collagen IV-conveyed signals can regulate chemokine production and promote liver metastasis. Oncogene 2018, 37, 3790–3805. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Brodt, P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin. Cancer Res. 2016, 22, 5971–5982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Reviews. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef]
- Milette, S.; Sicklick, J.K.; Lowy, A.M.; Brodt, P. Molecular Pathways: Targeting the Microenvironment of Liver Metastases. Clin. Cancer Res. 2017, 23, 6390–6399. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur J. Surg Oncol 2017, 43, 604–611. [Google Scholar] [CrossRef]
- Muto, A.; Gridelli, C. New immunotherapy in the treatment of advanced renal cancer. Expert Opin. Emerg. Drugs 2019. [Google Scholar] [CrossRef]
- Osmani, L.; Askin, F.; Gabrielson, E.; Li, Q.K. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin. Cancer Biol. 2018, 52, 103–109. [Google Scholar] [CrossRef]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Upadhrasta, S.; Zheng, L. Strategies in Developing Immunotherapy for Pancreatic Cancer: Recognizing and Correcting Multiple Immune “Defects” in the Tumor Microenvironment. J. Clin. Med. 2019, 8, 1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroldi, F.; Zaniboni, A. Immunotherapy for pancreatic cancer: Present and future. Immunotherapy 2017, 9, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Crudden, C.; Ilic, M.; Suleymanova, N.; Worrall, C.; Girnita, A.; Girnita, L. The dichotomy of the Insulin-like growth factor 1 receptor: RTK and GPCR: Friend or foe for cancer treatment? Growth Horm. IGF Res. 2015, 25, 2–12. [Google Scholar] [CrossRef]
- Girnita, L.; Worrall, C.; Takahashi, S.; Seregard, S.; Girnita, A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol. Life Sci. 2014, 71, 2403–2427. [Google Scholar] [CrossRef] [Green Version]
- Monteverde, M.; Milano, G.; Strola, G.; Maffi, M.; Lattanzio, L.; Vivenza, D.; Tonissi, F.; Merlano, M.; Nigro, C.L. The relevance of ADCC for EGFR targeting: A review of the literature and a clinically-applicable method of assessment in patients. Crit. Rev. Oncol./Hematol. 2015, 95, 179–190. [Google Scholar] [CrossRef]





{kind=link}
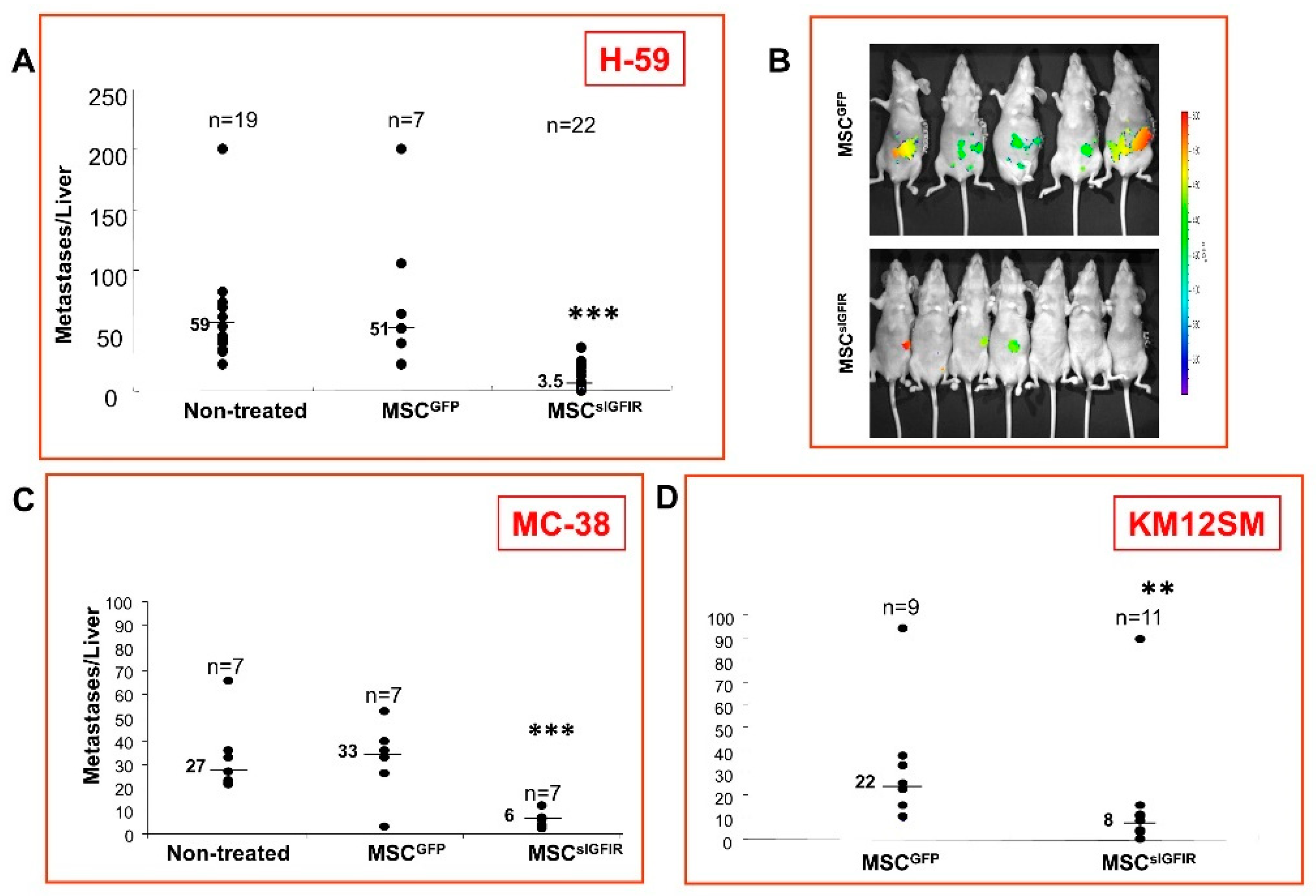{kind=link}
{kind=link}
{kind=link}
| Target | Approach | Advantages | Disadvantages | Reference |
|---|---|---|---|---|
| IGF- insulin receptor (IR) | Nucleic acid approach | High specificity via mRNA degradation | Toxicity, challenges in drug delivery and uptake Compensatory signaling through IR-A Low translational potential | [35] |
| Antibodies | Induce internalization and downregulation of IGF-IR | Adverse effects on glucose metabolism Hyperglycemia activation of IR-A by IGF-II nuclear translocation of IGF-IR Compensatory receptor tyrosine kinase (RTK) signaling | [36,37] | |
| Bispecific antibodies | Neutralizing two or more targets improved protein stability to oxidative and thermal stress Inhibit compensatory signaling by other RTKs | Steric hindrance large, reduced intra-tumoral penetration | [38,39] | |
| Tyrosine kinase inhibitors (TKI) | Cross reactivity with IR | Affects metabolic insulin signaling via IR-B hyperglycemia short half-life | [37,40] | |
| IGF- ligands | Antibodies | Block IGF-IR and IR-A activation Low affinity for insulin minimizes adverse effects on glucose metabolism Reduced ligand bioavailability in the serum | Efficacy depends on IGF-IR expression levels Reduced plasma IGF levels may trigger compensatory feedback mechanisms | [41] |
| Traps | Block IGF-IR and IR-A activation Low affinity for insulin minimizes adverse effects on glucose metabolism Reduce ligand bioavailability in the serum Fc fusion proteins increase serum half-life | Size may limit diffusion into the tumor site Oligomerization due to disulfide bonds may affect manufacturability Could potentially trigger a compensatory feedback mechanism upon long-term administration | [41,42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.M.; Qi, S.; Perrino, S.; Hashimoto, M.; Brodt, P. Targeting the IGF-Axis for Cancer Therapy: Development and Validation of an IGF-Trap as a Potential Drug. Cells 2020, 9, 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051098
Chen YM, Qi S, Perrino S, Hashimoto M, Brodt P. Targeting the IGF-Axis for Cancer Therapy: Development and Validation of an IGF-Trap as a Potential Drug. Cells. 2020; 9(5):1098. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051098
Chicago/Turabian StyleChen, Yinhsuan Michely, Shu Qi, Stephanie Perrino, Masakazu Hashimoto, and Pnina Brodt. 2020. "Targeting the IGF-Axis for Cancer Therapy: Development and Validation of an IGF-Trap as a Potential Drug" Cells 9, no. 5: 1098. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9051098