Proteins and Molecular Pathways Relevant for the Malignant Properties of Tumor-Initiating Pancreatic Cancer Cells

 , and
, and 
Abstract
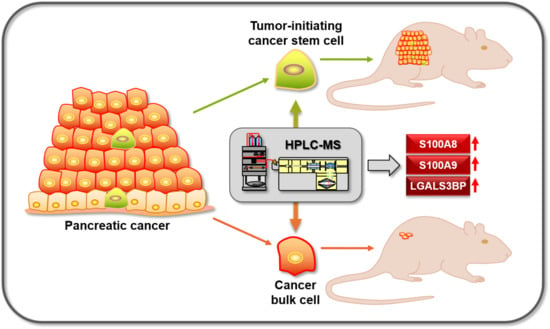
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Protein Extracts, Digestion, and Labeling
2.3. High-Performance Liquid Chromatography and Mass Spectrometry
2.4. Data Analysis
2.5. RNA Isolation and qPCR
2.6. shRNA and Migration Assay
2.7. Mouse Models
3. Results
3.1. Experimental Strategy for Proteomics and Functional Analysis
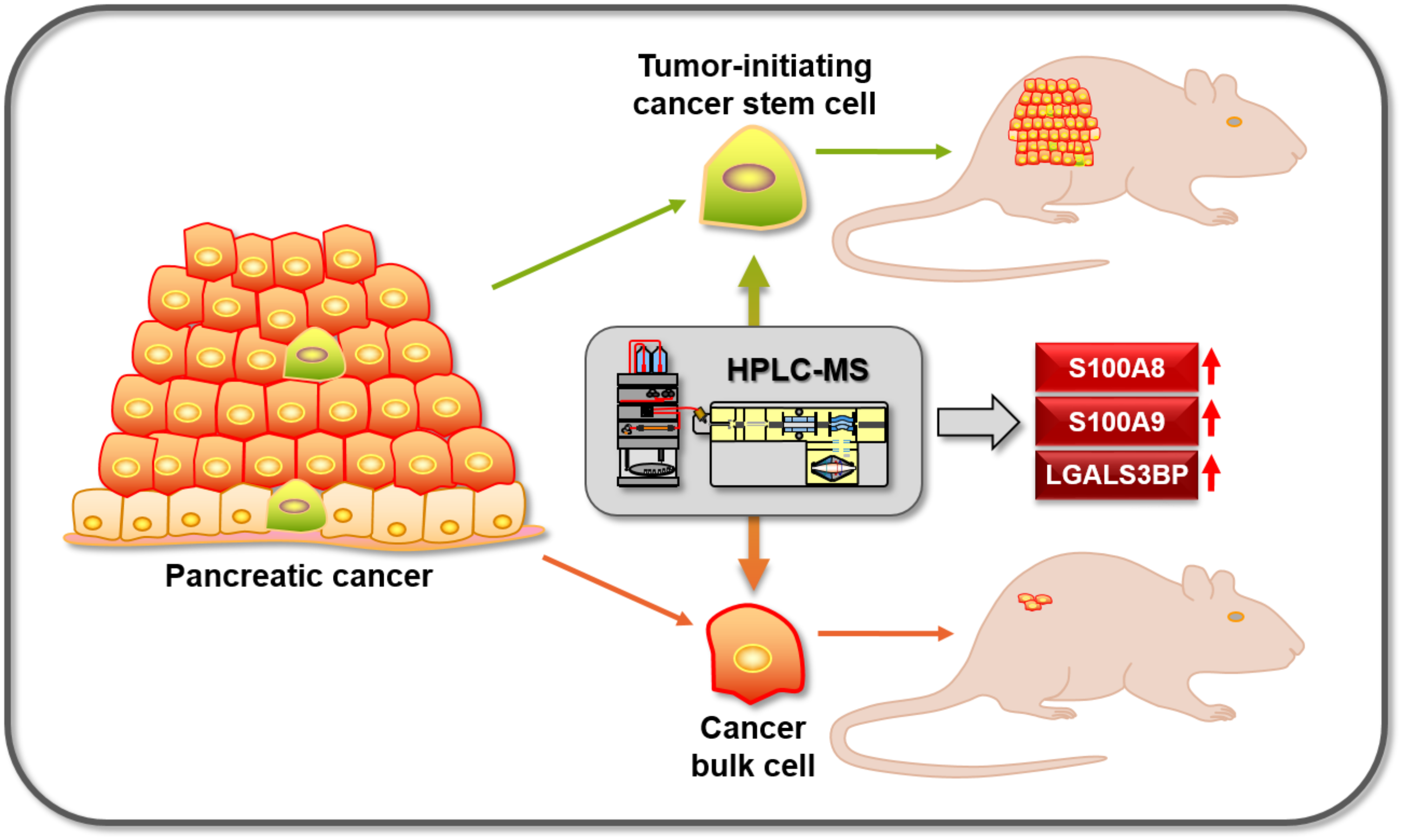
3.2. Differential Proteome, Gene Expression, and Pathway Analysis
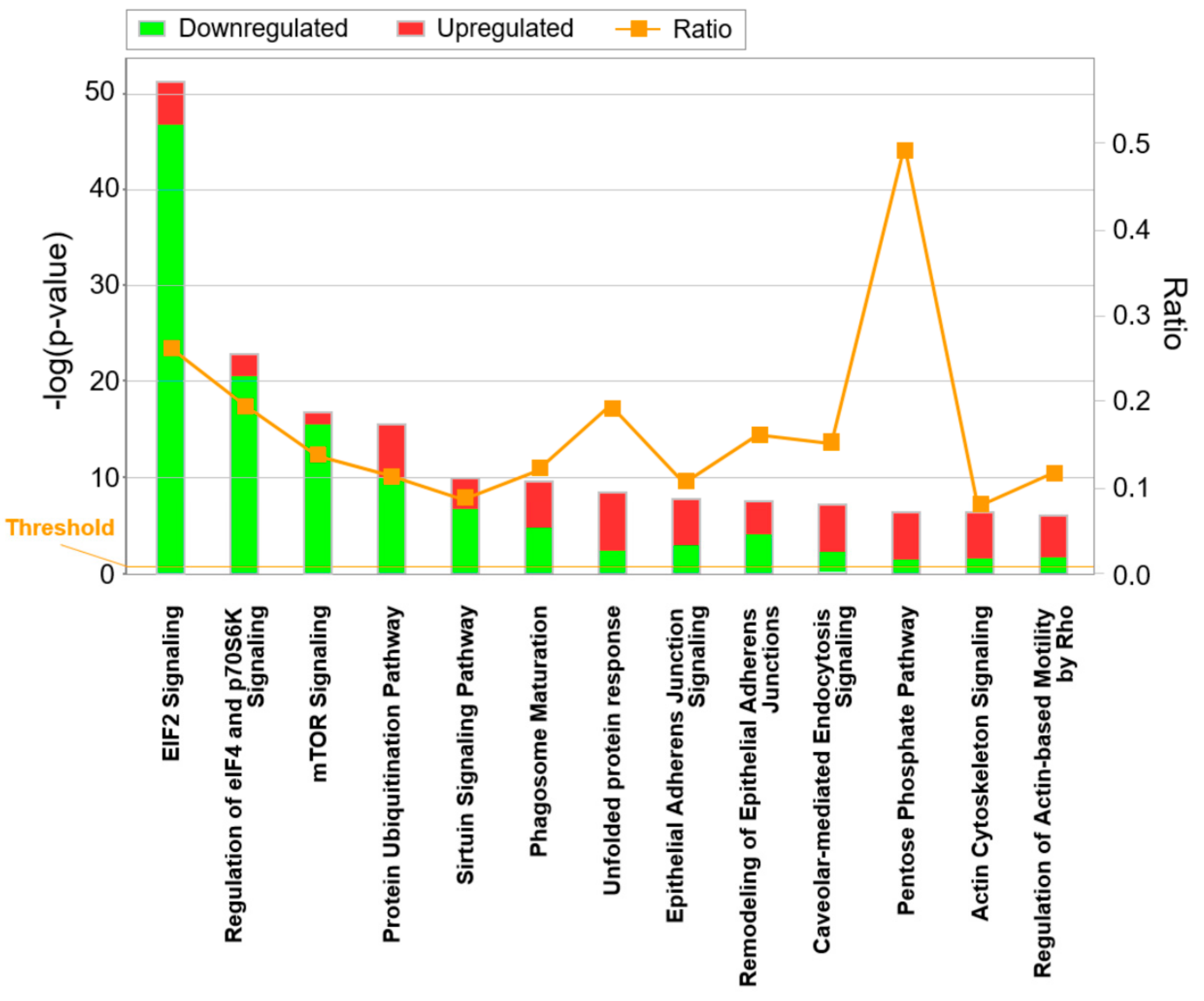
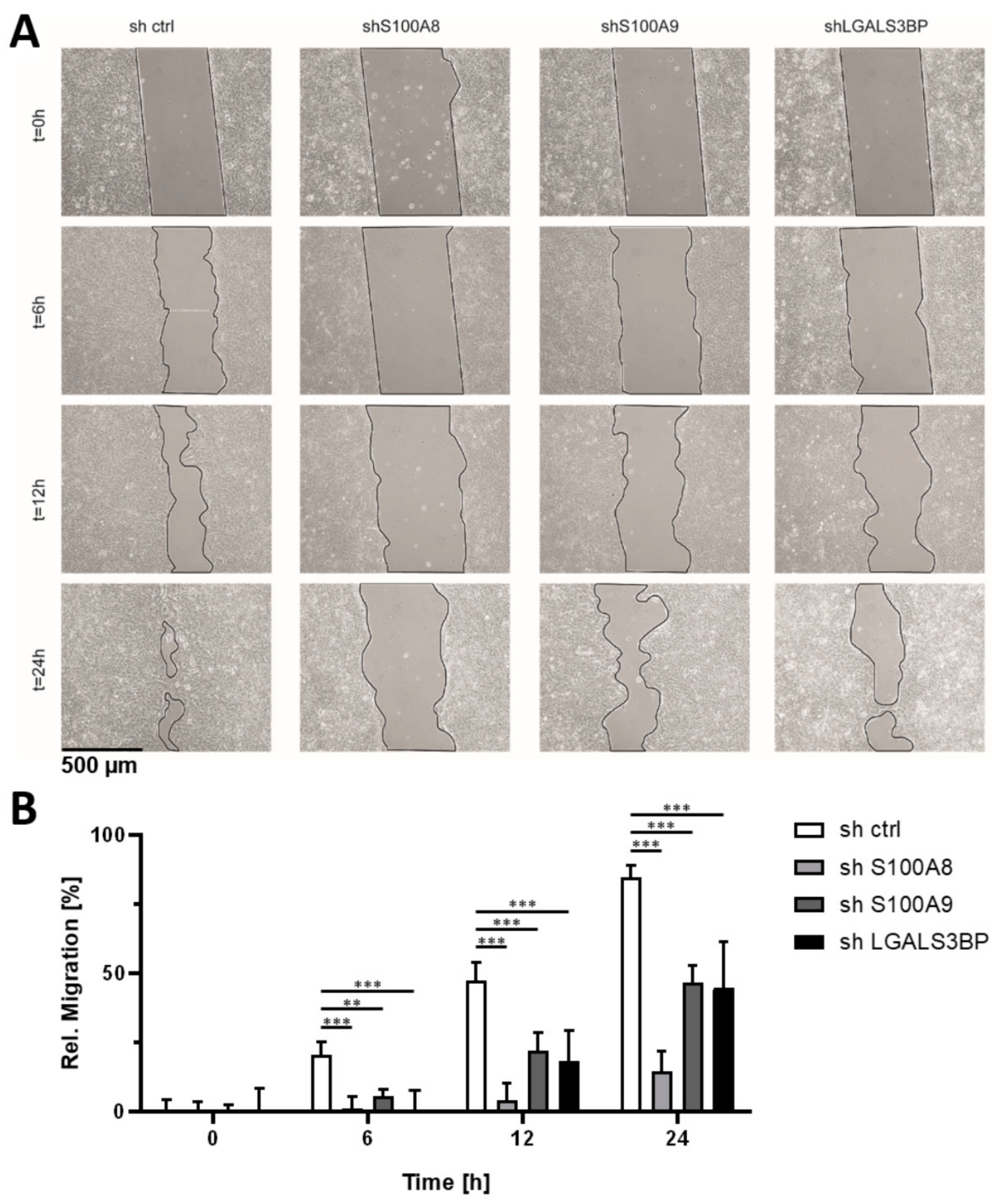
3.3. Candidate Verification by Functional Assays and Xenograft Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Kreso, A.; Dick, J. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Abstract SY28-02: Interactions between cancer stem cells and their niche govern metastatic colonization. Tumor Biology 2012, 72, SY28-02. [Google Scholar] [CrossRef]
- Blanpain, C. Tracing the cellular origin of cancer. Nature 2013, 15, 126–134. [Google Scholar] [CrossRef]
- Toh, T.B.; Lim, J.J.; Chow, E.K. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 29. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Valle, S.; Hijano, L.M.; Alcala, S.; Nocelo, M.A.; Sainz, B., Jr. The Ever-Evolving Concept of the Cancer Stem Cell in Pancreatic Cancer. Cancers 2018, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Cojoc, M.; Mäbert, K.; Muders, M.H.; Dubrovska, A. A role for cancer stem cells in therapy resistance: Cellular and molecular mechanisms. Semin. Cancer Biol. 2015, 31, 16–27. [Google Scholar] [CrossRef]
- Desai, A.; Yan, Y.; Gerson, S.L. Concise Reviews: Cancer Stem Cell Targeted Therapies: Toward Clinical Success. Stem Cells Transl. Med. 2018, 8, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Driessens, G.; Beck, B.; Caauwe, A.; Simons, B.; Blanpain, C. Defining the mode of tumour growth by clonal analysis. Nature 2012, 488, 527–530. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Michl, P.; Gress, T.M. Current concepts and novel targets in advanced pancreatic cancer. Gut 2012, 62, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Cao, H.; Le, D.; Yang, L.-X. Current status in chemotherapy for advanced pancreatic adenocarcinoma. Anticancer. Res. 2013, 33, 1785–1791. [Google Scholar]
- Heiler, S.; Wang, Z.; Zöller, M. Pancreatic cancer stem cell markers and exosomes–the incentive push. World J. Gastroenterol. 2016, 22, 5971–6007. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M.; Lepelletier, Y.; et al. Identification of Pancreatic Cancer Stem Cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.-M.; Koorstra, J.-B.; RajeshKumar, N.V.; He, X.; et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH Activity Selectively Defines an Enhanced Tumor-Initiating Cell Population Relative to CD133 Expression in Human Pancreatic Adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Di Magliano, M.P.; Simeone, D.M. c-Met Is a Marker of Pancreatic Cancer Stem Cells and Therapeutic Target. Gastroenterology 2011, 141, 2218–2227.e5. [Google Scholar] [CrossRef]
- Adikrisna, R.; Tanaka, S.; Muramatsu, S.; Aihara, A.; Ban, D.; Ochiai, T.; Irie, T.; Kudo, A.; Nakamura, N.; Yamaoka, S.; et al. Identification of Pancreatic Cancer Stem Cells and Selective Toxicity of Chemotherapeutic Agents. Gastroenterology 2012, 143, 234–245.e7. [Google Scholar] [CrossRef]
- Vaz, A.P.; Ponnusamy, M.P.; Rachagani, S.; Dey, P.; Ganti, A.K.; Batra, S.K. Novel role of pancreatic differentiation 2 in facilitating self-renewal and drug resistance of pancreatic cancer stem cells. Br. J. Cancer 2014, 111, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S.; Seshacharyulu, P.; Lakshmanan, I.; Vaz, A.P.; Chugh, S.; Sheinin, Y.M.; Mahapatra, S.; Batra, S.K.; Ponnusamy, M.P. hPaf1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells. Oncotarget 2017, 8, 14806–14820. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S.; Dey, P.; Vaz, A.P.; Bhaumik, S.R.; Ponnusamy, M.P.; Batra, S.K. PD2/PAF1 at the Crossroads of the Cancer Network. Cancer Res. 2018, 78, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Abel, E.; Simeone, D.M. Biology and Clinical Applications of Pancreatic Cancer Stem Cells. Gastroenterology 2013, 144, 1241–1248. [Google Scholar] [CrossRef]
- Penchev, V.R.; Rasheed, Z.A.; Maitra, A.; Matsui, W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 4277–4284. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin Signaling Drives Self-Renewal and Tumorigenicity of Pancreatic Cancer Stem Cells and Provides a Target for Combined Drug Therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Pan, S.; Brentnall, T.A.; Aebersold, R. Proteomic Profiling of Pancreatic Cancer for Biomarker Discovery. Mol. Cell. Proteom. 2005, 4, 523–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, C.; Pal, M.; Miller, F.R.; Barder, T.J.; Huber, C.G.; Lubman, D.M. Toward high sequence coverage of proteins in human breast cancer cells using on-line monolith-based HPLC-ESI-TOF MS compared to CE MS. Electrophoresis 2006, 27, 2126–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchior, K.; Tholey, A.; Heisel, S.; Keller, A.; Lenhof, H.-P.; Meese, E.; Huber, C.G. Proteomic Study of Human Glioblastoma Multiforme Tissue Employing Complementary Two-Dimensional Liquid Chromatography- and Mass Spectrometry-Based Approaches. J. Proteome Res. 2009, 8, 4604–4614. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Doondeea, J.B.; Gholami, A.M.; Janning, M.C.; Lemeer, S.; Kramer, K.; Eccles, S.A.; Gollin, S.; Grenman, R.; Walch, A.; et al. Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.; Imielinski, M.B.; Rejtar, T.; Richardson, E.A.; Thakur, D.; Sgroi, D.C.; Karger, B.L. In situ proteomic analysis of human breast cancer epithelial cells using laser capture microdissection: Annotation by protein set enrichment analysis and gene ontology. Mol. Cell. Proteom. 2010, 9, 2529–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.-C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhang, Y.; Tang, T.; Zhu, Q.; Shi, W.; Yin, X.; Xing, Y.; Shen, Y.; Pan, Y.; Jin, L. Transcriptome Profiling of Panc-1 Spheroid Cells with Pancreatic Cancer Stem Cells Properties Cultured by a Novel 3D Semi-Solid System. Cell. Physiol. Biochem. 2018, 47, 2109–2125. [Google Scholar] [CrossRef]
- Steiniger, S.C.; Coppinger, J.A.; Krüger, J.A.; Yates, J.R.; Janda, K.D. Quantitative mass spectrometry identifies drug targets in cancer stem cell-containing side population. Stem Cells 2008, 26, 3037–3046. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Li, C.; Shedden, K.A.; Lee, C.; Li, C.; Quoc, H.; Simeone, D.M.; Lubman, D.M. Quantitative Proteomic Profiling Studies of Pancreatic Cancer Stem Cells. J. Proteome Res. 2010, 9, 3394–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandi, J.; Dando, I.; Pozza, E.D.; Biondani, G.; Jenkins, R.; Elliott, V.; Park, K.; Fanelli, G.; Zolla, L.; Costello, E.; et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J. Proteom. 2017, 150, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In Vivo Selection and Characterization of Metastatic Variants from Human Pancreatic Adenocarcinoma by Using Orthotopic Implantation in Nude Mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premstaller, A.; Oberacher, H.; Huber, C.G. High-performance liquid chromatography-electrospray ionization mass spectrometry of single- and double-stranded nucleic acids using monolithic capillary columns. Anal. Chem. 2000, 72, 4386–4393. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Li, L. Exploring the Precursor Ion Exclusion Feature of Liquid Chromatography−Electrospray Ionization Quadrupole Time-of-Flight Mass Spectrometry for Improving Protein Identification in Shotgun Proteome Analysis. Anal. Chem. 2008, 80, 4696–4710. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; A Dianes, J.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, 11033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlbacher, O.; Reinert, K.; Gröpl, C.; Lange, E.; Pfeifer, N.; Schulz-Trieglaff, O.; Sturm, M. TOPP--the OpenMS proteomics pipeline. Bioinformatics 2007, 23, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Röst, H.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E.; et al. OpenMS: A flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 2016, 13, 741–748. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- The UniProt Consortium; UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [CrossRef] [Green Version]
- Tyanova, S.; Cox, J. Perseus: A Bioinformatics Platform for Integrative Analysis of Proteomics Data in Cancer Research. Advanced Structural Safety Studies 2018, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Leek, J.T.; Storey, J. Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3, e161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Regl, G.; Eichberger, T.; Frischauf, A.M.; Aberger, F. Efficient manipulation of Hedgehog/GLI signaling using retroviral expression systems. In Hedgehog Signaling Protocols; Humana Press: Totowa, NJ, USA, 2007; pp. 67–78. [Google Scholar]
- Faul, F.; Erdfelder, E.; Lang, A.-G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Faça, V.M.; Song, K.S.; Wang, H.; Zhang, Q.; Krasnoselsky, A.L.; Newcomb, L.F.; Plentz, R.R.; Gurumurthy, S.; Redston, M.S.; Pitteri, S.J.; et al. A Mouse to Human Search for Plasma Proteome Changes Associated with Pancreatic Tumor Development. PLoS Med. 2008, 5, e123. [Google Scholar] [CrossRef]
- Brandi, J.; Pozza, E.D.; Dando, I.; Biondani, G.; Robotti, E.; Jenkins, R.; Elliott, V.; Park, K.; Marengo, E.; Costello, E.; et al. Secretome protein signature of human pancreatic cancer stem-like cells. J. Proteom. 2016, 136, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Nie, S.; Wu, J.; Lubman, D.M. Target Proteomic Profiling of Frozen Pancreatic CD24+ Adenocarcinoma Tissues by Immuno-Laser Capture Microdissection and Nano-LC–MS/MS. J. Proteome Res. 2013, 12, 2791–2804. [Google Scholar] [CrossRef] [Green Version]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Lanfranca, M.P.; Thompson, J.; Bednar, F.; Halbrook, C.; Lyssiotis, C.; Levi, B.; Frankel, T.L. Metabolism and epigenetics of pancreatic cancer stem cells. Semin. Cancer Biol. 2019, 57, 19–26. [Google Scholar] [CrossRef]
- Hanahan, D.; A Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Srikrishna, G. S100A8 and S100A9: New insights into their roles in malignancy. J. Innate Immun. 2011, 4, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclerc, E.; Vetter, S.W. The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim. Biophys. Acta 2015, 1852, 2706–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Chitayat, S.; Hashemi, M.; Eshraghi, M.; Chazin, W.J.; Halayko, A.J.; Kerkhoff, C. S100A8/A9: A Janus-faced molecule in cancer therapy and tumorgenesis. Eur. J. Pharmacol. 2009, 625, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Vandal, K.; Rouleau, P.; Boivin, A.; Ryckman, C.; Talbot, M.; Tessier, P.A. Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J. Immunol. 2003, 171, 2602–2609. [Google Scholar] [CrossRef] [Green Version]
- Ang, C.W.; Nedjadi, T.; Sheikh, A.A.; Tweedle, E.M.; Tonack, S.; Honap, S.; Jenkins, R.E.; Park, B.; Schwarte-Waldhoff, I.; Azadeh, B.; et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis 2010, 31, 1541–1551. [Google Scholar] [CrossRef] [Green Version]
- Moon, A.; Yong, H.-Y.; Song, J.-I.; Cukovic, D.; Salagrama, S.; Kaplan, D.; Putt, D.; Kim, H.; Dombkowski, A.; Kim, H.-R.C. Global Gene Expression Profiling Unveils S100A8/A9 as Candidate Markers in H-Ras-Mediated Human Breast Epithelial Cell Invasion. Mol. Cancer Res. 2008, 6, 1544–1553. [Google Scholar] [CrossRef] [Green Version]
- Rafii, S.; Lyden, D. S100 chemokines mediate bookmarking of premetastatic niches. Nature 2006, 8, 1321–1323. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nature 2006, 8, 1369–1375. [Google Scholar] [CrossRef]
- Al-Ismaeel, Q.; Neal, C.P.; Al-Mahmoodi, H.; Almutairi, Z.; Al-Shamarti, I.; Straatman, K.; Jaunbocus, N.; Irvine, A.; Issa, E.; Moreman, C.; et al. ZEB1 and IL-6/11-STAT3 signalling cooperate to define invasive potential of pancreatic cancer cells via differential regulation of the expression of S100 proteins. Br. J. Cancer 2019, 121, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.J.; Chazin, W.J. High Level Expression and Dimer Characterization of the S100 EF-hand Proteins, Migration Inhibitory Factor-related Proteins 8 and 14. J. Biol. Chem. 1998, 273, 12427–12435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, J.A.R.; May, R.; Tanousis, K.; McNeill, E.; Mathies, M.; Gebhardt, C.; Henderson, R.; Robinson, M.J.; Hogg, N. Myeloid Cell Function in MRP-14 (S100A9) Null Mice. Mol. Cell. Biol. 2003, 23, 2564–2576. [Google Scholar] [CrossRef] [Green Version]
- Manitz, M.-P.; Horst, B.; Seeliger, S.; Strey, A.; Skryabin, B.V.; Gunzer, M.; Frings, W.; Schoönlau, F.; Roth, J.; Sorg, C.; et al. Loss of S100A9 (MRP14) Results in Reduced Interleukin-8-Induced CD11b Surface Expression, a Polarized Microfilament System, and Diminished Responsiveness to Chemoattractants In Vitro. Mol. Cell. Biol. 2003, 23, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Azizan, N.; Suter, M.A.; Liu, Y.; Logsdon, C.D. RAGE maintains high levels of NFκB and oncogenic Kras activity in pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 493, 592–597. [Google Scholar] [CrossRef]
- Shahab, U.; Ahmad, M.K.; Mahdi, A.A.; Waseem, M.; Arif, B.; Moinuddin; Ahmad, S. The receptor for advanced glycation end products: A fuel to pancreatic cancer. Semin. Cancer Biol. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Kasper, M.; Regl, G.; Frischauf, A.-M.; Aberger, F. GLI transcription factors: Mediators of oncogenic Hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, R.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Silverman, A.M.; Nakata, R.; Shimada, H.; Sposto, R.; Declerck, Y.A. A Galectin-3-Dependent Pathway Upregulates Interleukin-6 in the Microenvironment of Human Neuroblastoma. Cancer Res. 2012, 72, 2228–2238. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, E.; Tinari, N.; Semeraro, D.; Traini, S.; Fichera, I.; Cumashi, A.; La Sorda, R.; Spinella, F.; Bagnato, A.; Lattanzio, R.; et al. LGALS3BP, lectin galactoside-binding soluble 3 binding protein, induces vascular endothelial growth factor in human breast cancer cells and promotes angiogenesis. J. Mol. Med. 2012, 91, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Woodman, N.; Pinder, S.E.; Tajadura, V.; Le Bourhis, X.; Gillett, C.; Delannoy, P.; Burchell, J.; Julien, S. Two E-selectin ligands, BST-2 and LGALS3BP, predict metastasis and poor survival of ER-negative breast cancer. Int. J. Oncol. 2016, 49, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, R.; Yoshimaru, T.; Matsushita, Y.; Matsuo, T.; Ono, M.; Park, J.; Sasa, M.; Miyoshi, Y.; Nakamura, Y.; Katagiri, T. The GALNT6-LGALS3BP axis promotes breast cancer cell growth. Int. J. Oncol. 2019, 56, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Künzli, B.M.; Berberat, P.O.; Zhu, Z.W.; Martignoni, M.; Kleeff, J.; Tempia-Caliera, A.A.; Fukuda, M.; Zimmermann, A.; Friess, H.; Büchler, M.W. Influences of the lysosomal associated membrane proteins (Lamp-1, Lamp-2) and Mac-2 binding protein (Mac-2-BP) on the prognosis of pancreatic carcinoma. Cancer 2002, 94, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5’–3’) | Reverse (5’–3’) |
|---|---|---|
| RPLP0 | GGCACCATTGAAATCCTGAGTGATGTG | TTGCGGACACCCTCCAGGAAGC |
| S100A8 | TTGACCGAGCTGGAGAAAGCCTTG | GGTCATCCCTGTAGACGGCATGGA |
| S100A9 | GGGAATTCAAAGAGCTGGTGCGAA | TCTTCTCGTGGGAGGCCCAGGTTA |
| LGALS3BP | CCTGGGCTGGCTGAAGAGCAACT | GCCCCGCTGGCTGTCAAAGA |
| NDRG1 | AATGACTCGTTACCTGCCGCCCATC | CCCCGATCCCCGACTTTTCTACTCA |
| CTSD | GGGCGAGTACATGATCCCCTGTGAG | CTTCCCGGCCTGCGACACCTT |
| ALDH3A1 | CATGTTCTCCAGCAACGACAAGGTGA | AGGCAAGAGCGGCGGTGAGAGA |
| CD9 | GTTTGGCTGGGGGCGTGGAA | TGCGCCGATGATGTGGAATTTATTGTC |
| ARPC3 | TCGACCCTCAGAATGATAAACCCAGCA | CGGGCTCCCTTCACTGTCCAGGT |
| Target Gene | Sigma Mission shRNA Clone ID |
|---|---|
| Control | shctrl (shc002 (scrambled non-target shRNA)) |
| S100A8 | TRCN0000053777 |
| S100A9 | TRCN0000053803 |
| LGALS3BP | TRCN0000029414 |
| S100A9 | S100A8 | LGALS3BP | NDRG1 | CTSD | ALDH3A1 | CD9 | ARPC3 | |
|---|---|---|---|---|---|---|---|---|
| PTX a) | ++ | ++ | ++ | + | + | + | + | + |
| qPCR a) | +++++ | +++++ | +++ | ++++ | 0 | ++ | + | + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samonig, L.; Loipetzberger, A.; Blöchl, C.; Rurik, M.; Kohlbacher, O.; Aberger, F.; Huber, C.G. Proteins and Molecular Pathways Relevant for the Malignant Properties of Tumor-Initiating Pancreatic Cancer Cells. Cells 2020, 9, 1397. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061397
Samonig L, Loipetzberger A, Blöchl C, Rurik M, Kohlbacher O, Aberger F, Huber CG. Proteins and Molecular Pathways Relevant for the Malignant Properties of Tumor-Initiating Pancreatic Cancer Cells. Cells. 2020; 9(6):1397. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061397
Chicago/Turabian StyleSamonig, Lisa, Andrea Loipetzberger, Constantin Blöchl, Marc Rurik, Oliver Kohlbacher, Fritz Aberger, and Christian G. Huber. 2020. "Proteins and Molecular Pathways Relevant for the Malignant Properties of Tumor-Initiating Pancreatic Cancer Cells" Cells 9, no. 6: 1397. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061397