RIG-I Plays a Dominant Role in the Induction of Transcriptional Changes in Zika Virus-Infected Cells, which Protect from Virus-Induced Cell Death

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Generation of Knock-Out Cells
2.3. ZIKV
2.4. IFNβ ELISA
2.5. Type I IFN Bioassay
2.6. IFNβ Promoter Reporter Assay
2.7. Caspase Activity Assay
2.8. qRT-PCR
2.9. 3′mRNA Sequencing
2.10. FACS
2.11. Western Blot Analysis
3. Results
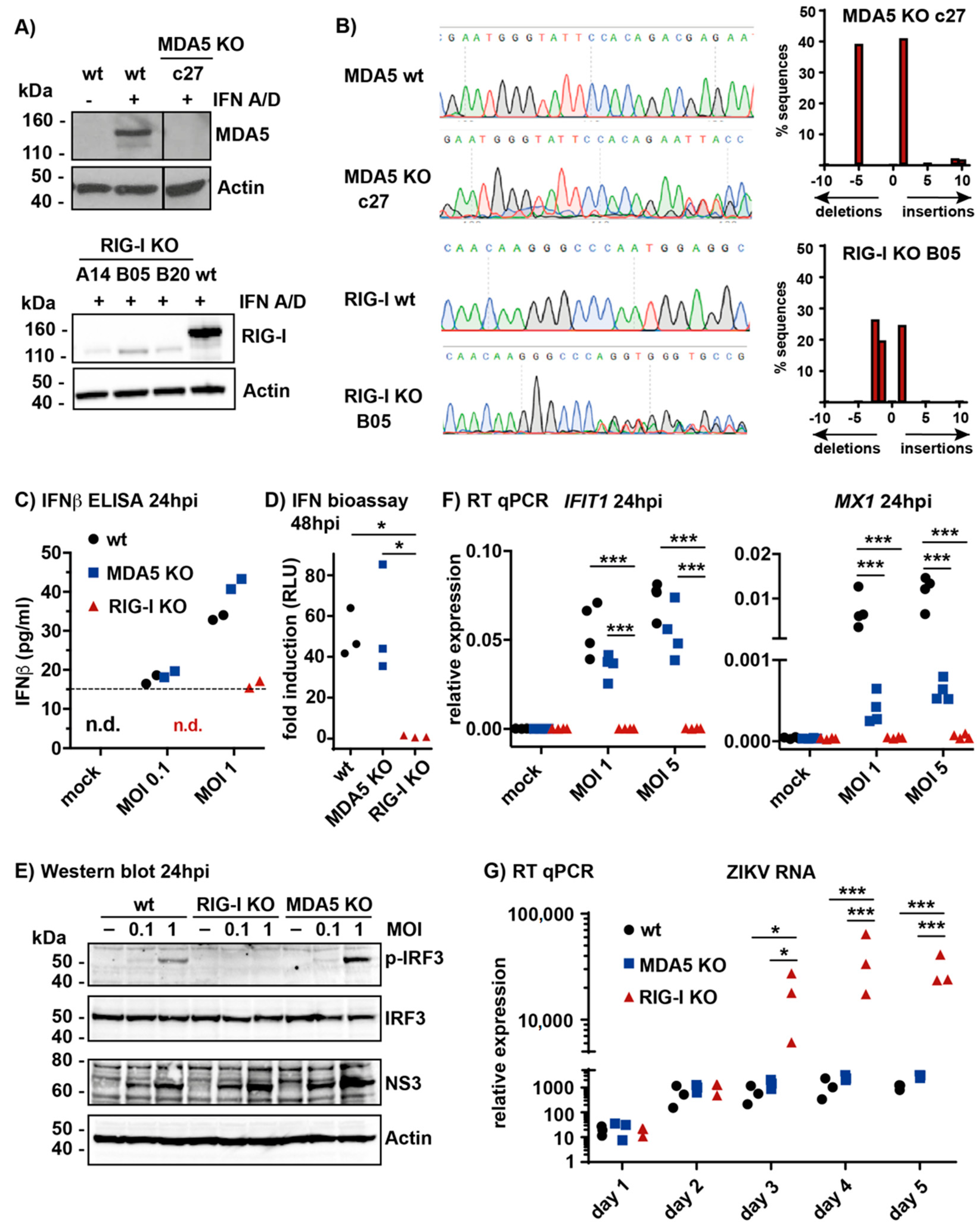
3.1. RIG-I Is the Main Sensor for ZIKV Infection in A549 Cells
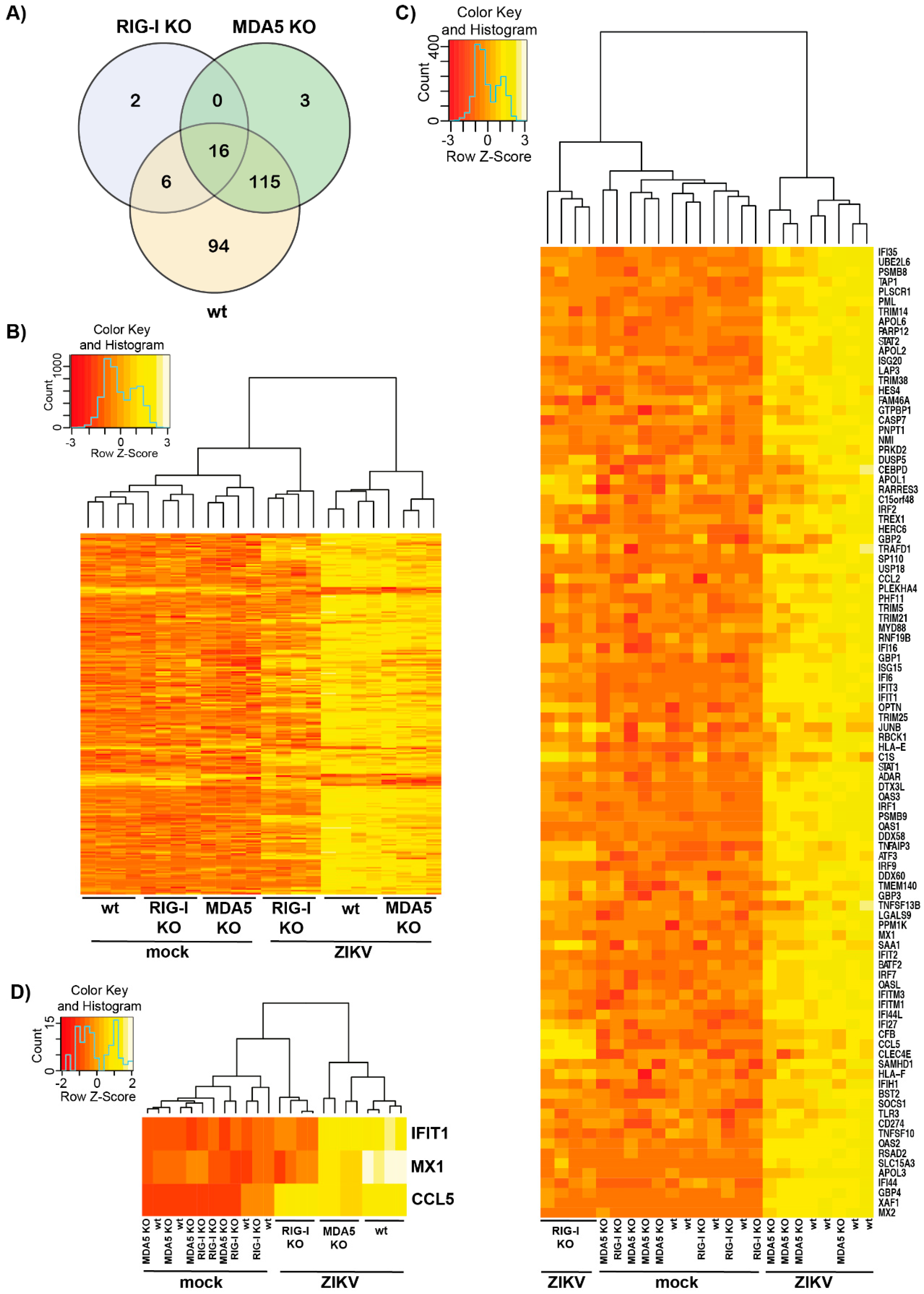
3.2. Transcriptomic Analysis of ZIKV-Infected Cells Indicates that RIG-I Plays a Dominant Role in ISG Induction
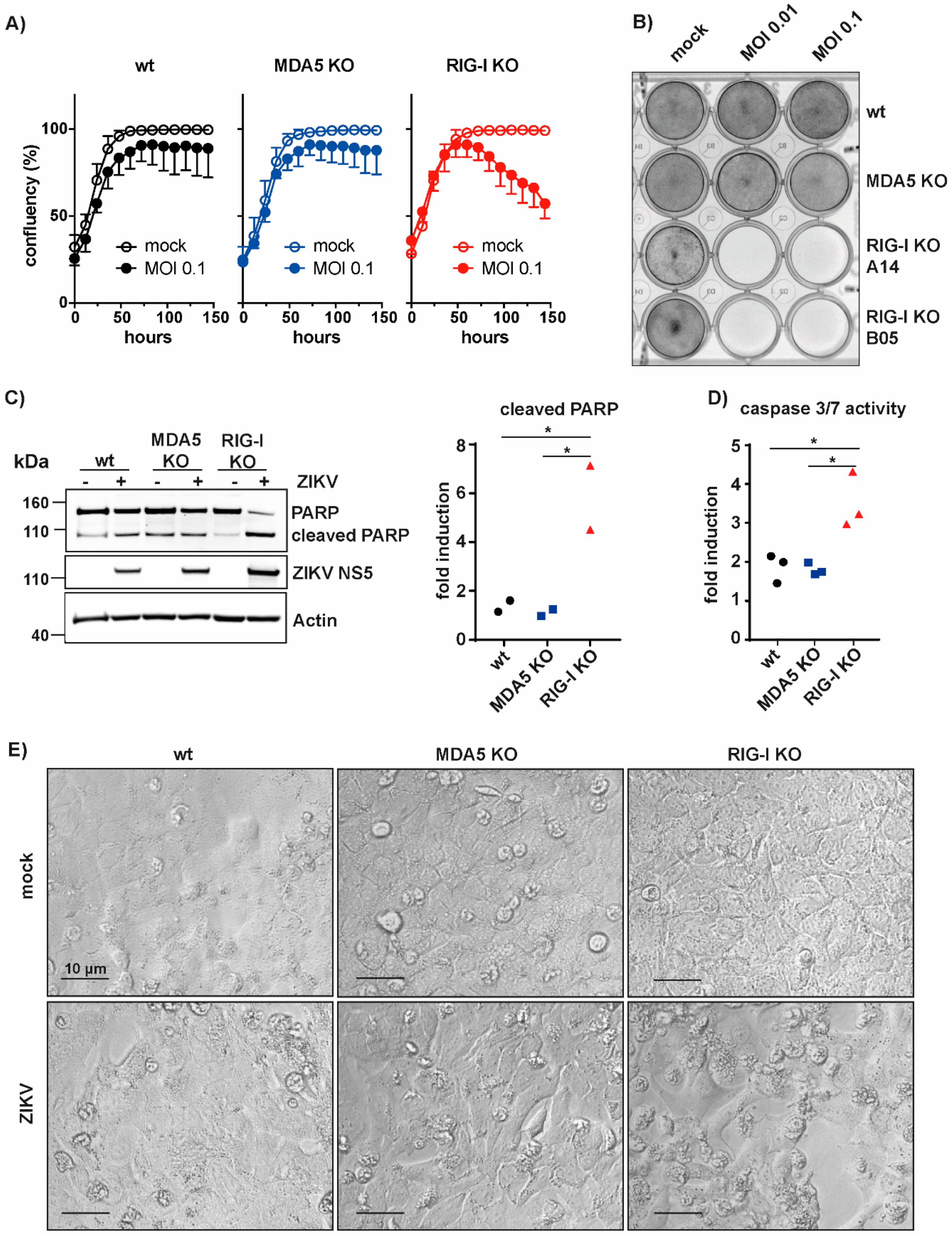
3.3. RIG-I-Mediated Signaling Protects A549 Cells from Apoptosis
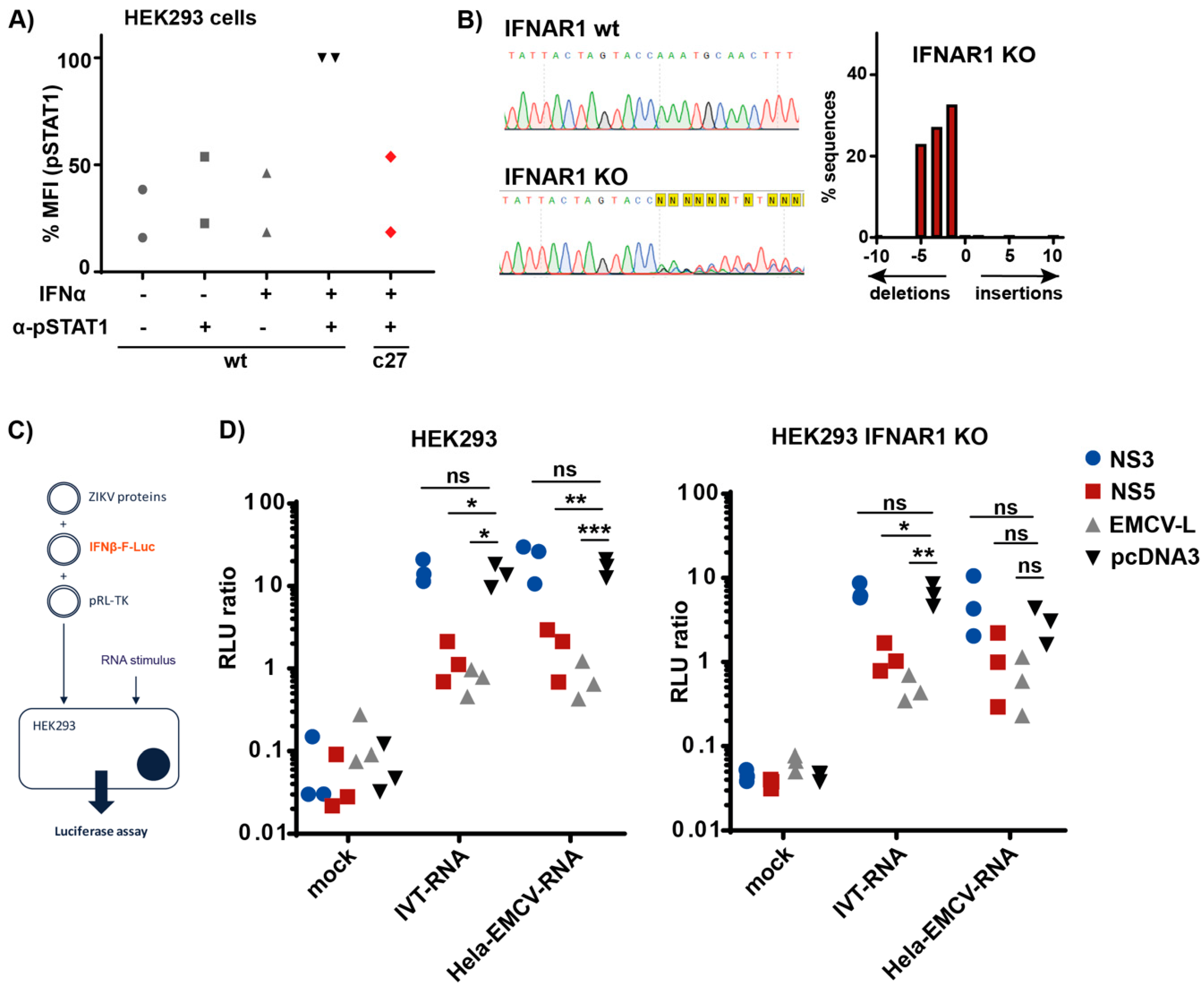
3.4. ZIKV NS5 Inhibits Type I IFN Induction
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faria, N.R.; Azevedo, R.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.Y.; Shi, W.F.; Qin, C.F. The evolution of Zika virus from Asia to the Americas. Nat. Rev. Microbiol. 2019, 17, 131–139. [Google Scholar] [CrossRef]
- Li, H.; Saucedo-Cuevas, L.; Shresta, S.; Gleeson, J.G. The Neurobiology of Zika Virus. Neuron 2016, 92, 949–958. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barre Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar] [CrossRef]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256.e1244. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Li, S.; Ma, S.; Jia, L.; Zhang, F.; Zhang, Y.; Zhang, J.; Wong, G.; Zhang, S.; Lu, X.; et al. Zika Virus Causes Testis Damage and Leads to Male Infertility in Mice. Cell 2016, 167, 1511–1524.e10. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.; Balasubramaniam, V.R.; Brown, J.A.; Mena, I.; Grant, A.; Bardina, S.V.; Maringer, K.; Schwarz, M.C.; Maestre, A.M.; Sourisseau, M.; et al. A novel Zika virus mouse model reveals strain specific differences in virus pathogenesis and host inflammatory immune responses. PLoS Pathog. 2017, 13, e1006258. [Google Scholar] [CrossRef] [Green Version]
- Hertzog, J.; Dias Junior, A.G.; Rigby, R.E.; Donald, C.L.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Ketkar, H.; Geng, T.; Lo, E.; Wang, L.; Xi, J.; Sun, Q.; Zhu, Z.; Cui, Y.; Yang, L.; et al. Zika Virus Non-structural Protein 4A Blocks the RLR-MAVS Signaling. Front. Microbiol. 2018, 9, 1350. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Dong, X.; He, Z.; Wu, Y.; Zhang, S.; Lin, J.; Yang, Y.; Chen, J.; An, S.; Yin, Y.; et al. Zika virus antagonizes interferon response in patients and disrupts RIG-I-MAVS interaction through its CARD-TM domains. Cell Biosci. 2019, 9, 46. [Google Scholar] [CrossRef]
- Esser-Nobis, K.; Aarreberg, L.D.; Roby, J.A.; Fairgrieve, M.R.; Green, R.; Gale, M., Jr. Comparative analysis of African and Asian lineage-derived Zika Virus strains reveals differences in activation of and sensitivity to antiviral innate immunity. J. Virol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Riedl, W.; Acharya, D.; Lee, J.H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M.U. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503.e6. [Google Scholar] [CrossRef]
- Liao, X.; Xie, H.; Li, S.; Ye, H.; Li, S.; Ren, K.; Li, Y.; Xu, M.; Lin, W.; Duan, X.; et al. 2′, 5′-Oligoadenylate Synthetase 2 (OAS2) Inhibits Zika Virus Replication through Activation of Type Iota IFN Signaling Pathway. Viruses 2020, 12, 418. [Google Scholar] [CrossRef] [Green Version]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.S.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef] [PubMed]
- Roby, J.A.; Esser-Nobis, K.; Dewey-Verstelle, E.C.; Fairgrieve, M.R.; Schwerk, J.; Lu, A.Y.; Soveg, F.W.; Hemann, E.A.; Hatfield, L.D.; Keller, B.C.; et al. Flavivirus Nonstructural Protein NS5 Dysregulates HSP90 to Broadly Inhibit JAK/STAT Signaling. Cells 2020, 9, 899. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Cumberworth, S.L.; Clark, J.J.; Kohl, A.; Donald, C.L. Inhibition of type I interferon induction and signalling by mosquito-borne flaviviruses. Cell Microbiol. 2017, 19. [Google Scholar] [CrossRef] [Green Version]
- Best, S.M. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signaling. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira Franca, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef]
- Hilton, L.; Moganeradj, K.; Zhang, G.; Chen, Y.H.; Randall, R.E.; McCauley, J.W.; Goodbourn, S. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 2006, 80, 11723–11732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, A.; Maelfait, J.; Davenne, T.; Partridge, T.; Peng, Y.; Mayer, A.; Dong, T.; Kaever, V.; Borrow, P.; Rehwinkel, J. Viruses transfer the antiviral second messenger cGAMP between cells. Science 2015, 349, 1228–1232. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 21 October 2019).
- Flohr, F.; Schneider-Schaulies, S.; Haller, O.; Kochs, G. The central interactive region of human MxA GTPase is involved in GTPase activation and interaction with viral target structures. FEBS Lett. 1999, 463, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Choi, H.C.; Chun, Y.H.; Kim, H.; Park, S.H. Characterization of chromosomal aberrations in lung cancer cell lines by cross-species color banding. Cancer Genet. Cytogenet. 2001, 124, 62–70. [Google Scholar] [CrossRef]
- Isaka, T.; Nestor, A.L.; Takada, T.; Allison, D.C. Chromosomal variations within aneuploid cancer lines. J. Histochem. Cytochem. 2003, 51, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Ames, H.M.; Tipton, A.; Vezina, G.; Liu, J.S.; Scafidi, J.; Torii, M.; Rodriguez, F.J.; du Plessis, A.; DeBiasi, R.L. Differential neuronal susceptibility and apoptosis in congenital Zika virus infection. Ann. Neurol. 2017, 82, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.R.; Moreli, J.B.; Marques, R.E.; Papa, M.P.; Meuren, L.M.; Rahal, P.; de Arruda, L.B.; Oliani, A.H.; Oliani, D.; Oliani, S.M.; et al. Zika-virus-infected human full-term placental explants display pro-inflammatory responses and undergo apoptosis. Arch. Virol. 2018, 163, 2687–2699. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liao, H.M.; Li, B.; Tsai, S.; Hung, G.C.; Lo, S.C. Comparative Genomics, Infectivity and Cytopathogenicity of American Isolates of Zika Virus that Developed Persistent Infections in Human Embryonic Kidney (HEK293) Cells. Int. J. Mol. Sci. 2019, 20, 3035. [Google Scholar] [CrossRef] [Green Version]
- Sherer, M.L.; Khanal, P.; Talham, G.; Brannick, E.M.; Parcells, M.S.; Schwarz, J.M. Zika virus infection of pregnant rats and associated neurological consequences in the offspring. PLoS ONE 2019, 14, e0218539. [Google Scholar] [CrossRef]
- Anfasa, F.; Goeijenbier, M.; Widagdo, W.; Siegers, J.Y.; Mumtaz, N.; Okba, N.; van Riel, D.; Rockx, B.; Koopmans, M.P.G.; Meijers, J.C.M.; et al. Zika Virus Infection Induces Elevation of Tissue Factor Production and Apoptosis on Human Umbilical Vein Endothelial Cells. Front. Microbiol. 2019, 10, 817. [Google Scholar] [CrossRef]
- Li, G.; Bos, S.; Tsetsarkin, K.A.; Pletnev, A.G.; Despres, P.; Gadea, G.; Zhao, R.Y. The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity. Viruses 2019, 11, 157. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, Q.; Li, X.; Qiu, Z.; Li, A.; Liang, W.; Chen, H.; Cai, X.; Chen, X.; Duan, X.; et al. Zika Virus Envelope Protein induces G2/M Cell Cycle Arrest and Apoptosis via an Intrinsic Cell Death Signaling Pathway in Neuroendocrine PC12 Cells. Int. J. Biol. Sci. 2018, 14, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, J.R.; Azevedo, R.S.S.; Martins Filho, A.J.; Araujo, M.T.F.; Moutinho, E.R.C.; Baldez Vasconcelos, B.C.; Cruz, A.C.R.; Oliveira, C.S.; Martins, L.C.; Baldez Vasconcelos, B.H.; et al. Correlation between Apoptosis and in Situ Immune Response in Fatal Cases of Microcephaly Caused by Zika Virus. Am. J. Pathol. 2018, 188, 2644–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nature Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, S.; He, J.; Guest, J.D.; Ma, Z.; Yang, L.; Pierce, B.G.; Tang, Q.; Zhang, Y.J. Zika virus NS5 protein antagonizes type I interferon production via blocking TBK1 activation. Virology 2018, 527, 180–187. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Lin, Y.C.; Boone, M.; Meuris, L.; Lemmens, I.; Van Roy, N.; Soete, A.; Reumers, J.; Moisse, M.; Plaisance, S.; Drmanac, R.; et al. Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 2014, 5, 4767. [Google Scholar] [CrossRef] [Green Version]
- Bylund, L.; Kytola, S.; Lui, W.O.; Larsson, C.; Weber, G. Analysis of the cytogenetic stability of the human embryonal kidney cell line 293 by cytogenetic and STR profiling approaches. Cytogenet. Genome Res. 2004, 106, 28–32. [Google Scholar] [CrossRef]
- Hato, S.V.; Ricour, C.; Schulte, B.M.; Lanke, K.H.; de Bruijni, M.; Zoll, J.; Melchers, W.J.; Michiels, T.; van Kuppeveld, F.J. The mengovirus leader protein blocks interferon-alpha/beta gene transcription and inhibits activation of interferon regulatory factor 3. Cell. Microbiol. 2007, 9, 2921–2930. [Google Scholar] [CrossRef]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Lorière, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I Recognizes the 5′ Region of Dengue and Zika Virus Genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.; Bartenschlager, R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013, 2, 32–48. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.; Shresta, S.; Gleeson, J.G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, E.; Ramani, A.; Karow, U.; Gottardo, M.; Natarajan, K.; Gooi, L.M.; Goranci-Buzhala, G.; Krut, O.; Peters, F.; Nikolic, M.; et al. Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids. Cell Stem Cell 2017, 20, 397–406.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El-Kalamouni, C.; Nativel, B.; Rondeau, P.; Misse, D.; Gadea, G.; Viranaicken, W.; Despres, P. The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-beta production and apoptosis induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef]
- Vielle, N.J.; Zumkehr, B.; Garcia-Nicolas, O.; Blank, F.; Stojanov, M.; Musso, D.; Baud, D.; Summerfield, A.; Alves, M.P. Silent infection of human dendritic cells by African and Asian strains of Zika virus. Sci. Rep. 2018, 8, 5440. [Google Scholar] [CrossRef] [Green Version]
- Turpin, J.; Frumence, E.; Despres, P.; Viranaicken, W.; Krejbich-Trotot, P. The ZIKA Virus Delays Cell Death Through the Anti-Apoptotic Bcl-2 Family Proteins. Cells 2019, 8, 1338. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, R.; Melen, K.; Westenius, V.; Jiang, M.; Osterlund, P.; Khan, H.; Vapalahti, O.; Julkunen, I.; Kakkola, L. Zika Virus Non-Structural Protein NS5 Inhibits the RIG-I Pathway and Interferon Lambda 1 Promoter Activation by Targeting IKK Epsilon. Viruses 2019, 11, 1024. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Wang, W.; Wang, Y.; Chen, K.; Xiao, F.; Hu, D.; Hui, L.; Liu, W.; Feng, Y.; Li, G.; et al. NS5 Conservative Site Is Required for Zika Virus to Restrict the RIG-I Signaling. Front. Immunol. 2020, 11, 51. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tao, M.; Han, W.; Fan, Z.; Imran, M.; Cao, S.; Ye, J. Nuclear localization of Zika virus NS5 contributes to suppression of type I interferon production and response. J. Gen. Virol. 2019. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schilling, M.; Bridgeman, A.; Gray, N.; Hertzog, J.; Hublitz, P.; Kohl, A.; Rehwinkel, J. RIG-I Plays a Dominant Role in the Induction of Transcriptional Changes in Zika Virus-Infected Cells, which Protect from Virus-Induced Cell Death. Cells 2020, 9, 1476. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061476
Schilling M, Bridgeman A, Gray N, Hertzog J, Hublitz P, Kohl A, Rehwinkel J. RIG-I Plays a Dominant Role in the Induction of Transcriptional Changes in Zika Virus-Infected Cells, which Protect from Virus-Induced Cell Death. Cells. 2020; 9(6):1476. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061476
Chicago/Turabian StyleSchilling, Mirjam, Anne Bridgeman, Nicki Gray, Jonny Hertzog, Philip Hublitz, Alain Kohl, and Jan Rehwinkel. 2020. "RIG-I Plays a Dominant Role in the Induction of Transcriptional Changes in Zika Virus-Infected Cells, which Protect from Virus-Induced Cell Death" Cells 9, no. 6: 1476. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061476