Transcriptomic microRNA Profiling of Dendritic Cells in Response to Gut Microbiota-Secreted Vesicles

 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Isolation of Membrane Vesicles
2.3. Generation and Stimulation of Human Monocyte-Derived DCs
2.4. Flow Cytometry Analysis of Surface Markers
2.5. RNA Isolation
2.6. Small RNA-seq Library Preparation and Sequencing
2.7. Small RNA-seq Pre-Processing and Differential Expression Analysis
2.8. Gene Ontology Enrichment Analysis of miRNA Targets
2.9. Validation of miRNAs and Predicted mRNA Targets by Quantitative Reverse Transcription-PCR (RT-qPCR)
2.10. Cytokine Quantification
2.11. Statistical Analysis
2.12. Ethics
3. Results
3.1. miRNA Expression Profiling
3.2. Differential Expression of miRNAs in Response to Probiotic and Commensal-Derived MVs
3.3. Selection of miRNAs
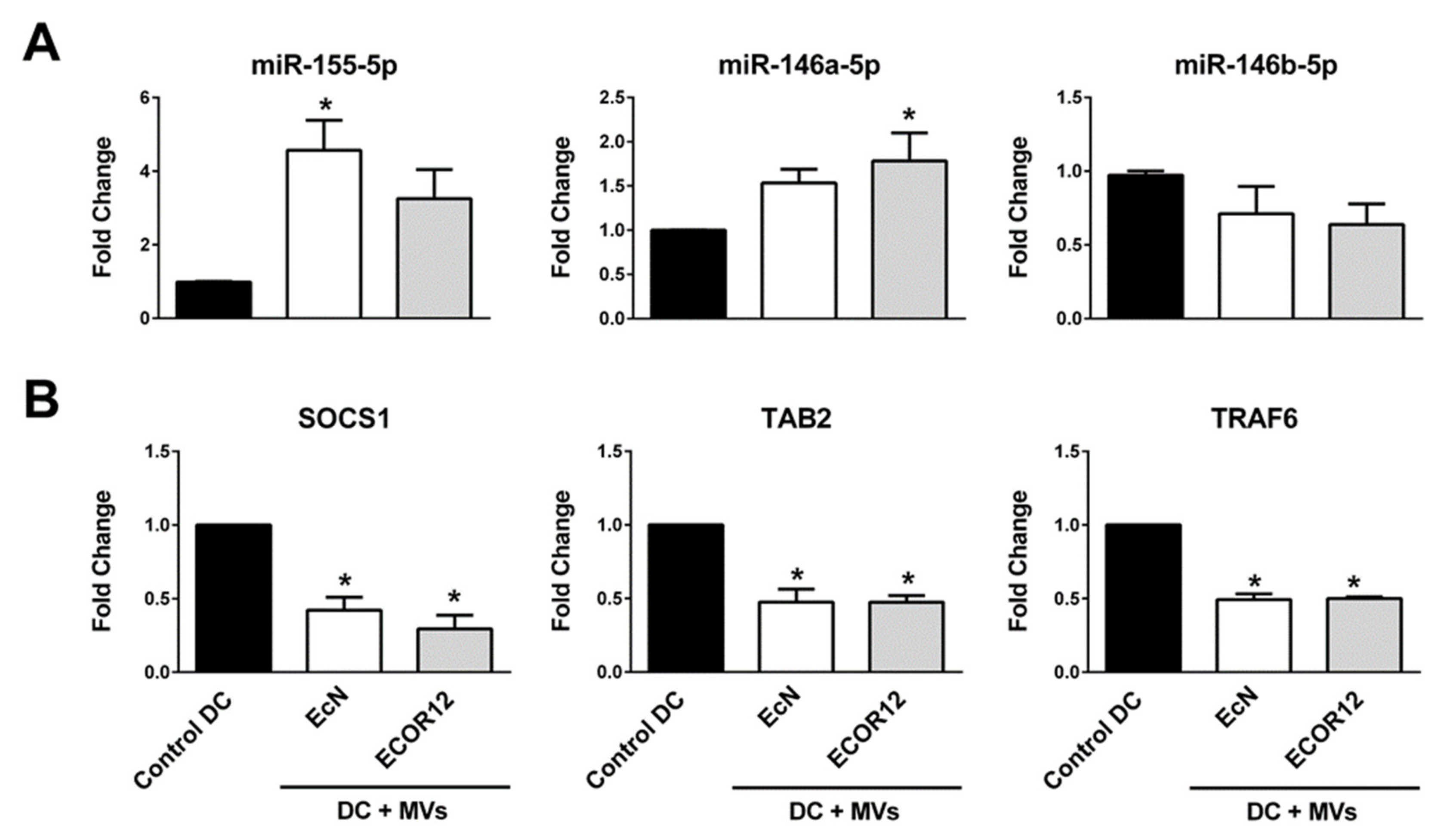
3.4. Validation of Selected miRNAs
3.5. Expression Analysis of Selected miRNA Target Genes Relevant for DC Maturation and Function
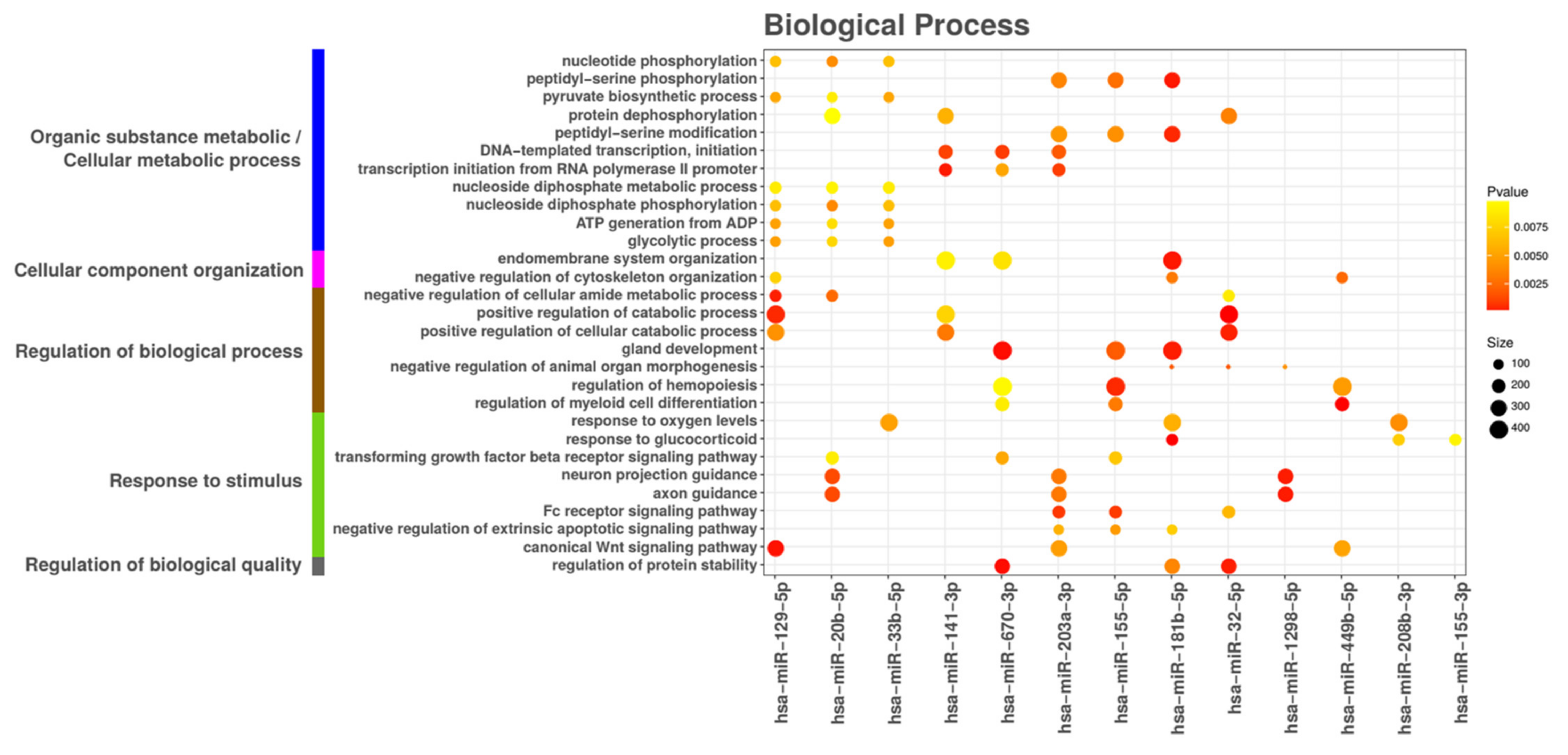
3.6. miRNA Target Functional Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, N.D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Caballero, S.; Pamer, E.G. Microbiota-mediated inflammation and antimicrobial defense in the intestine. Annu. Rev. Immunol. 2015, 33, 227–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rescigno, M.; Lopatin, U.; Chieppa, M. Interactions among dendritic cells, macrophages, and epithelial cells in the gut: Implications for immune tolerance. Curr. Opin. Immunol. 2008, 20, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakawa, H.O.; Tomari, Y. The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015, 25, 651–665. [Google Scholar] [CrossRef]
- Masotti, A. Interplays between gut microbiota and gene expression regulation by miRNAs. Front. Cell. Infect. Microbiol. 2012, 2, 137. [Google Scholar] [CrossRef] [Green Version]
- Runtsch, M.C.; Round, J.L.; O’Connell, R.M. MicroRNAs and the regulation of intestinal homeostasis. Front. Genet. 2014, 5, 347. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, C.; Mano, M.; Eulalio, A. MicroRNAs at the host-bacteria interface: Host defense or bacterial offense. Trends Microbiol. 2018, 27, 206–218. [Google Scholar] [CrossRef]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [Green Version]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, B.; Urdaci, M.C.; Margolles, A. Extracellular proteins secreted by probiotic bacteria as mediators of effects that promote mucosa-bacteria interactions. Microbiology 2010, 156, 3232–3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Huang, S.; Wang, Y.; Cai, S.; Yu, H.; Liu, H.; Zeng, X.; Zhang, G.; Qiao, S. Bridging intestinal immunity and gut microbiota by metabolites. Cell. Mol. Life Sci. 2019, 76, 3917–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Ren, Y.; Fu, X. Inter-kingdom signaling between gut microbiota and their host. Cell. Mol. Life Sci. 2019, 76, 2383–2389. [Google Scholar] [CrossRef]
- Guerrero-Mandujano, A.; Hernández-Cortez, C.; Ibarra, J.A.; Castro-Escarpulli, G. The outer membrane vesicles: Secretion system type zero. Traffic 2017, 18, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Kaparakis-Liaskos, M.; Ferrero, R.L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015, 15, 375–387. [Google Scholar] [CrossRef]
- Aguilera, L.; Toloza, L.; Giménez, R.; Odena, A.; Oliveira, E.; Aguilar, J.; Badia, J.; Baldomà, L. Proteomic analysis of outer membrane vesicles from the probiotic strain Escherichia coli Nissle 1917. Proteomics 2014, 14, 222–229. [Google Scholar] [CrossRef]
- Zakharzhevskaya, N.B.; Vanyushkina, A.A.; Altukhov, I.A.; Shavarda, A.L.; Butenko, I.O.; Rakitina, D.V.; Nikitina, A.S.; Manolov, A.I.; Egorova, A.N.; Kulikov, E.E.; et al. Outer membrane vesicles secreted by pathogenic and nonpathogenic Bacteroides fragilis represent different metabolic activities. Sci. Rep. 2017, 7, 5008. [Google Scholar] [CrossRef]
- Cañas, M.A.; Giménez, R.; Fábrega, M.J.; Toloza, L.; Baldomà, L.; Badia, J. Outer membrane vesicles from the probiotic Escherichia coli Nissle 1917 and the commensal ECOR12 enter intestinal epithelial cells via clathrin-dependent endocytosis and elicit differential effects on DNA damage. PLoS ONE 2016, 11, e0160374. [Google Scholar] [CrossRef]
- Alvarez, C.S.; Badia, J.; Bosch, M.; Giménez, R.; Baldomà, L. Outer membrane vesicles and soluble factors released by probiotic Escherichia coli Nissle 1917 and commensal ECOR63 enhance barrier function by regulating expression of tight junction proteins in intestinal epithelial cells. Front. Microbiol. 2016, 7, 1981. [Google Scholar] [CrossRef] [Green Version]
- Cañas, M.A.; Fábrega, M.J.; Giménez, R.; Badia, J.; Baldomà, L. Outer membrane vesicles from probiotic and commensal Escherichia coli activate Nod1-mediated immune responses in intestinal epithelial cells. Front. Microbiol. 2018, 9, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fábrega, M.J.; Aguilera, L.; Giménez, R.; Varela, E.; Cañas, M.A.; Antolín, M.; Badia, J.; Baldomà, L. Activation of immune and defense responses in the intestinal mucosa by outer membrane vesicles of commensal and probiotic Escherichia coli strains. Front. Microbiol. 2016, 7, 705. [Google Scholar] [CrossRef] [Green Version]
- Fábrega, M.J.; Rodríguez-Nogales, A.; Garrido-Mesa, J.; Algieri, F.; Badia, J.; Giménez, R.; Gálvez, J.; Baldomà, L. Intestinal anti-inflammatory effects of outer membrane vesicles from Escherichia coli Nissle 1917 in DSS-experimental colitis in mice. Front. Microbiol. 2017, 8, 1274. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garrido, N.; Fábrega, M.J.; Vera, R.; Giménez, R.; Badia, J.; Baldomà, L. Membrane vesicles from the probiotic Nissle 1917 and gut resident Escherichia coli strains distinctly modulate human dendritic cells and subsequent T cell responses. J. Funct. Foods 2019, 61, 103495. [Google Scholar] [CrossRef]
- Ochman, H.; Selander, R.K. Standard reference strains of Escherichia coli from natural populations. J. Bacteriol. 1984, 157, 690–693. [Google Scholar] [CrossRef] [Green Version]
- Fast QC: A Quality Control Tool for High Throughput Sequence Data (FastQC: v0.11.7). Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 March 2020).
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2015, 15, 182. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008 (R: v3.5.1). Available online: https://www.R-project.org/ (accessed on 20 March 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: New York, NY, USA, 2016. [Google Scholar]
- Ggplot2 Create Elegant Data Visualisations Using the Grammar of Graphics (ggplot2:v3.0.1). Available online: https://ggplot2.tidyverse.org/reference/ (accessed on 20 March 2020).
- Raivo, K. Pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: https://cran.r-project.org/package=pheatmap (accessed on 20 March 2020).
- Chen, H. VennDiagram: Generate High-Resolution Venn and Euler Plots. R Package Version 1.6.20. 2018. Available online: https://cran.r-project.org/package=VennDiagram (accessed on 20 March 2020).
- Chen, Y.; Wang, X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chou, C.H.; Shrestha, S.; Yang, C.D.; Chang, N.W.; Lin, Y.L.; Liao, K.W.; Huang, W.C.; Sun, T.-H.; Tu, S.-J.; Lee, W.-H.; et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018, 46, D296–D302. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- The Gene Ontology Consortium. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [CrossRef] [Green Version]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: a web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Lind, E.F.; Millar, D.G.; Dissanayake, D.; Savage, J.C.; Grimshaw, N.K.; Kerr, W.G.; Ohashi, P.S. MiR-155 upregulation in dendritic cells is sufficient to break tolerance in vivo by negatively regulating SHIP1. J. Immunol. 2015, 195, 4632–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, L.A.; Boardman, D.A.; Tung, S.L.; Lechler, R.; Lombardi, G. MicroRNAs affect dendritic cell function and phenotype. Immunology 2014, 144, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Liu, F.; Jia, H.; Zhang, Q.; Ying, L.; Liu, W.; Liu, H.; Yu, B.; Wu, J. Inhibition of microRNA let-7i depresses maturation and functional state of dendritic cells in response to lipopolysaccharide stimulation via targeting suppressor of cytokine signaling 1. J. Immunol. 2011, 187, 1674–1683. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Huang, X.; Lu, C.; Cairo, M.S.; Zhou, X. MicroRNA-146a and microRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J. Biol. Chem. 2015, 290, 2831–2841. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; He, Y.S.; Wang, X.Q.; Lu, L.; Chen, Q.J.; Sun, Z.; Shen, W.F. MiR-146a inhibits oxidized low-density lipoprotein-induced lipid accumulation and inflammatory response via targeting toll-like receptor 4. FEBS Lett. 2011, 585, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Curtale, G.; Renzi, T.A.; Mirolo, M.; Drufuca, L.; Albanese, M.; De Luca, M.C.; Rossato, M.; Bazzoni, F.; Locati, M. Multi-Step Regulation of the TLR4 Pathway by the miR-125a~99b~let-7e Cluster. Front. Immunol. 2018, 9, 2037. [Google Scholar] [CrossRef] [Green Version]
- Tili, F.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; He, H.T.; Yang, Q. MiR-589-5p is a potential prognostic marker of hepatocellular carcinoma and regulates tumor cell growth by targeting MIG-6. Neoplasma 2018, 65, 753–761. [Google Scholar] [CrossRef]
- Han, S.Y.; Han, H.B.; Tian, X.Y.; Sun, H.; Xue, D.; Zhao, C.; Jiang, S.T.; He, X.R.; Zheng, W.X.; Wang, J.; et al. MicroRNA-33a-3p suppresses cell migration and invasion by directly targeting PBX3 in human hepatocellular carcinoma. Oncotarget 2016, 7, 42461–42473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabanelli, S.; Očadlíková, D.; Ciciarello, M.; Salvestrini, V.; Lecciso, M.; Jandus, C.; Metz, R.; Evangelisti, C.; Laury-Kleintop, L.; Romero, P.; et al. The SOCS3-independent expression of IDO2 supports the homeostatic generation of T regulatory cells by human dendritic cells. J. Immunol. 2014, 192, 1231–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hocès de la Guardia, A.; Staedel, C.; Kaafarany, I.; Clément, A.; Roubaud-Baudron, C.; Mégraud, F.; Lehours, P. Inflammatory cytokine and microRNA responses of primary human dendritic cells cultured with Helicobacter pylori strains; eCollection 2013. Front. Microbiol. 2013, 4, 236. [Google Scholar] [CrossRef] [Green Version]
- Pascale, A.; Marchesi, N.; Marelli, C.; Coppola, A.; Luzi, L.; Govoni, S.; Giustina, A.; Gazzaruso, C. Microbiota and metabolic diseases. Endocrine 2018, 61, 357–371. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two key modulators of immune response and tumor development. Non-Coding RNA 2017, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Evel-Kabler, K.; Strube, R.; Chen, S.Y. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat. Biotechnol. 2004, 22, 1546–1553. [Google Scholar] [CrossRef]
- Pauls, S.D.; Marshall, A.J. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur J. Immunol. 2017, 47, 932–945. [Google Scholar] [CrossRef]
- Dueck, A.; Eichner, A.; Sixt, M.; Meister, G. A miR-155-dependent microRNA hierarchy in dendritic cell maturation and macrophage activation. FEBS Lett. 2014, 588, 632–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahid, M.A.; Pauley, K.M.; Satoh, M.; Chan, E.K. miR-146a is critical for endotoxin-induced tolerance. Implication in innate immunity. J. Biol. Chem 2009, 284, 34590–34599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc. Nat. Acad. Sci. USA 2013, 110, 11499–11504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, A.R.; Fordham, J.B.; Ganesh, B.; Naresa, S. miR-24, miR-30b and miR-142-3p interfere with antigen processing and presentation by primary macrophages and dendritic cells. Sci. Rep. 2016, 6, 32925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metz, R.; Smith, C.; DuHadaway, J.B.; Chandler, P.; Baban, B.; Merlo, L.M.; Pigott, E.; Keough, M.P.; Rust, S.; Mellor, A.L.; et al. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int. Immunol. 2014, 26, 357–367. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene a | Sequence | Gene Accession Number |
|---|---|---|
| PBX3 | F: CAAGTCGGAGCCAATGTG R: ATGTAGCTCAGGGAAAAGTG | NM_001134778.2 |
| MIG6 | F: CTACTGGAGCAGTCGCAGTG R: CCTCTTCATGTGGTCCCAAG | AJ276373.1 |
| TNF-α | F: CTTCTGCCTGCTG CACTTTGGA R: TCCCAAAGTAGACCTGCCCAGA | NM_000594.4 |
| TRAF6 | F: CCTTTGGCAAATGTCATCTGTG R: CTCTGCATCTTTTCATGGCAAC | NM_145803.3 |
| TLR4 | F: ATATTGACAGGAAACCCCATCCA R: AGAGAGATTGAGTAGGGGCATTT | NM_003266.4 |
| IRAK1 | F: TCAGAACGGCTTCTACTGCCTG R: TACCCAGAAGGATGTCCAGTCG | NM_001569.4 |
| SOCS1 | F: TTGCCTGGAACCATGTGG R: GGTCCTGGCCTCCAGATACAG | NM_003745.1 |
| SHIP1 | F: GCTGGAGGAA GAGGACACAG R: AGTCAGCGGGATGTTTCTTG | NM_001017915.3 |
| TAB2 | F: CAGCCTGGTCCCTGGACTACT R: ATGAATGGTTGGTGGTTGTGAA | NM_015093.5 |
| IFN-γ | F: TGACCAGAGCATCCAAAAGA R: CTCTTCGACCTCGAAACAGC | NM_000619.3 |
| IDO2 | F: AGAAGTGGGCTTTGCTCTGC R: TGGCAAGACCTTACGGACATCTC | NM_002164.6 |
| HPRT-1 | F: CCTGGCGTCGTGATTAGTGAT R: AGACGTTCAGTCCTGTCCATAA | NM_000194 |
| Group/ Sample | GEO Accession Number | # Reads | # Reads after Trimming | # Uniquely Mapped Reads | % Uniquely Mapped Reads | # Multi- Mapped Reads | % Multi- Mapped Reads |
|---|---|---|---|---|---|---|---|
| Control-1 | GSM4286621 | 43.177.375 | 39.844.522 | 33.379.572 | 83.77 | 163.725 | 0.41 |
| Control-2 | GSM4286622 | 42.027.544 | 41.121.701 | 28.037.324 | 68.18 | 323.871 | 0.79 |
| Control-3 | GSM4286623 | 42.035.628 | 40.198.491 | 32.221.679 | 80.16 | 213.212 | 0.53 |
| EcN-MVs-1 | GSM4286624 | 41.710.678 | 40.507.274 | 32.961.507 | 81.37 | 181.397 | 0.45 |
| EcN-MVs-2 | GSM4286625 | 37.508.894 | 34.316.470 | 26.739.425 | 77.92 | 183.188 | 0.53 |
| EcN-MVs-3 | GSM4286626 | 34.990.472 | 33.402.664 | 27.717.617 | 82.98 | 117.631 | 0.35 |
| ECOR12-MVs-1 | GSM4286627 | 15.384.378 | 15.125.212 | 12.702.801 | 83.98 | 44.291 | 0.29 |
| ECOR12-MVs-2 | GSM4286628 | 19.955.575 | 19.682.866 | 16.032.149 | 81.45 | 60.202 | 0.31 |
| ECOR12-MVs-3 | GSM4286629 | 20.245.746 | 20.008.948 | 16.915.467 | 84.54 | 53.680 | 0.27 |
| Expression | miRNa | DC + ECOR12 MVs vs. Control | DC + EcN MVs vs. Control | ||||
|---|---|---|---|---|---|---|---|
| Fold Change | log2 Fold Change | Padj | Fold Change | log2 Fold Change | Padj | ||
| Common UP EcN ECOR12 | hsa-miR-155-5p | 10.93 | 3.45 | 5.23 × 10−115 | 10.34 | 3.37 | 7.1 × 10−110 |
| hsa-let-7i-3p | 2.83 | 1.5 | 2.84 × 10−25 | 2.97 | 1.57 | 1.83 × 10−28 | |
| hsa-miR-146a-5p | 7.89 | 2.98 | 1.89 × 10−44 | 1.79 | 0.84 | 3.5 × 10−4 | |
| UP ECOR12 NOT EcN | hsa-miR-146b-5p | 5.86 | 2.55 | 3.34 × 10−30 | 0.92 | (−0.10) | 8.2 × 10−1 |
| hsa-miR-24-3p | 5.98 | 2.58 | 3.2 × 10−25 | 1.41 | 0.5 | 1.25 × 10−1 | |
| hsa-miR-125b-5p | 3.23 | 1.69 | 2.5 × 10−6 | (−0.23) | 0.85 | 7.3 x 10−1 | |
| hsa-miR-125a-5p | 1.91 | 0.93 | 1.74 × 10−7 | 1.15 | 0.2 | 4.67 × 10−1 | |
| hsa-miR-99b-5p | 2.36 | 1.24 | 1.66 × 10−5 | 1.06 | 0.08 | 8.99 × 10−1 | |
| hsa-let-7e-5p | 1.97 | 0.98 | 3.33 × 10−9 | 1.23 | 0.30 | 1.57 × 10−1 | |
| hsa-miR-589-5p | 1.82 | 0.86 | 3.33 × 10−9 | −0.62 | (−0.69) | 3.96 × 10−6 | |
| UP EcN NOT ECOR12 | hsa-miR-33a-3p | −0.54 | (−0.95) | 1.58 × 10−6 | 1.84 | 0.88 | 7.45 × 10−8 |
| hsa-miR-29a-5p | −0.76 | (−0.4) | 8.49 × 10−2 | 1.54 | 0.62 | 1.49 × 10−3 | |
| Target | miRNA | References |
|---|---|---|
| SHIP1 | miR-155-5p | [49] |
| SOCS1 | miR-155-5p, let-7i-3p | [50,51] |
| TAB2 | miR-155-5p | [50] |
| TRAF6 | miR-146a-5p, miR-146b-5p | [52] |
| TLR4 | miR-146a-5p, miR125a-5p | [53,54] |
| IFN-γ | miR-24-3p | MIRDB and TargetScan Vent |
| TNF-α | miR-125a-5p, let-7e-5p, mir125-5p | [54,55] |
| IL-12 | miR-125a-5p, let-7e-5p | [54] |
| IDO2 | miR-29a-5p | MIRDB and TargetScan Vent |
| MIG6 | miR-589-5p | [56] |
| PBX3 | miR-33a-3p | [57] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Garrido, N.; Bonnin, S.; Riera, M.; Gíménez, R.; Badia, J.; Baldomà, L. Transcriptomic microRNA Profiling of Dendritic Cells in Response to Gut Microbiota-Secreted Vesicles. Cells 2020, 9, 1534. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061534
Díaz-Garrido N, Bonnin S, Riera M, Gíménez R, Badia J, Baldomà L. Transcriptomic microRNA Profiling of Dendritic Cells in Response to Gut Microbiota-Secreted Vesicles. Cells. 2020; 9(6):1534. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061534
Chicago/Turabian StyleDíaz-Garrido, Natalia, Sarah Bonnin, Marta Riera, Rosa Gíménez, Josefa Badia, and Laura Baldomà. 2020. "Transcriptomic microRNA Profiling of Dendritic Cells in Response to Gut Microbiota-Secreted Vesicles" Cells 9, no. 6: 1534. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061534