Amyloid Proteins and Peripheral Neuropathy
by
 and
and
Mohammed M. H. Albariqi
1,2, 
Sjoukje Engelsman
1,
Niels Eijkelkamp
1,*,† and
Jo W. M. Höppener
1,3,† 1
Center for Translational Immunology, University Medical Center Utrecht, Utrecht University, 3584 EA Utrecht, The Netherlands
2
The National Centre for Genomic Technology, Life Science and Environment Research Institute, King Abdulaziz City for Science and Technology, P.O. Box 6086, 11461 Riyadh, Saudi Arabia
3
Center for Molecular Medicine, University Medical Center Utrecht, Utrecht University, 3584 EA Utrecht, The Netherlands
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(6), 1553; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061553
Submission received: 2 June 2020
/
Revised: 20 June 2020
/
Accepted: 22 June 2020
/
Published: 26 June 2020
(This article belongs to the Special Issue Novel Insights into Molecular Mechanisms of Chronic Pain)
Abstract
:Painful peripheral neuropathy affects millions of people worldwide. Peripheral neuropathy develops in patients with various diseases, including rare familial or acquired amyloid polyneuropathies, as well as some common diseases, including type 2 diabetes mellitus and several chronic inflammatory diseases. Intriguingly, these diseases share a histopathological feature—deposits of amyloid-forming proteins in tissues. Amyloid-forming proteins may cause tissue dysregulation and damage, including damage to nerves, and may be a common cause of neuropathy in these, and potentially other, diseases. Here, we will discuss how amyloid proteins contribute to peripheral neuropathy by reviewing the current understanding of pathogenic mechanisms in known inherited and acquired (usually rare) amyloid neuropathies. In addition, we will discuss the potential role of amyloid proteins in peripheral neuropathy in some common diseases, which are not (yet) considered as amyloid neuropathies. We conclude that there are many similarities in the molecular and cell biological defects caused by aggregation of the various amyloid proteins in these different diseases and propose a common pathogenic pathway for “peripheral amyloid neuropathies”.
1. Introduction
The nervous system and the endocrine system are major regulatory systems that maintain homeostasis of the body in response to both endogenous and environmental stimuli. The peripheral nervous system encompasses all neurological tissues outside of the brain and spinal cord, including 12 pairs of cranial nerves and 31 pairs of spinal nerves, along with their roots and branches [1]. Peripheral sensory neurons are vulnerable to damage because they may have very long axons and the sensory neurons in the dorsal root ganglia have an attenuated protective neurovascular barrier compared with the blood–brain barrier and blood–nerve barrier. Damage to peripheral nerves causes peripheral neuropathy [1], which can involve gain- or loss-of-function. Gain-of-function symptoms include increased pain sensitivity [2], whereas loss-of-function symptoms include hyposensitivity and even a total loss of sensation (numbness) [3,4].
Neuropathic pain affects 7–10% of the population [5]. Peripheral neuropathic pain may have multiple causes, including diabetes mellitus (DM), hereditary disorders, inflammation and infections, autoimmune diseases, kidney failure, chronic alcoholism, and certain medications—especially those used to treat cancer and HIV/AIDS [6,7]. Although various diseases or causes may be at the root of peripheral neuropathy, the precise molecular mechanisms that cause (painful) peripheral neuropathy are still not well understood [7].
Diseases caused by amyloid—extracellular deposits of aggregated, misfolded proteins—are known as amyloidoses [8,9]. Amyloidosis can be inherited or acquired. Some types of amyloidoses are life threatening, such as immunoglobulin light-chain amyloidosis (AL) and serum amyloid A protein amyloidosis (SAA) [10,11]. Intriguingly, among patients with amyloidosis, peripheral neuropathy is observed in a substantial number, and for some types of amyloidosis this occurs in all patients, pointing towards amyloid being a cause of peripheral neuropathy (see Table 1).
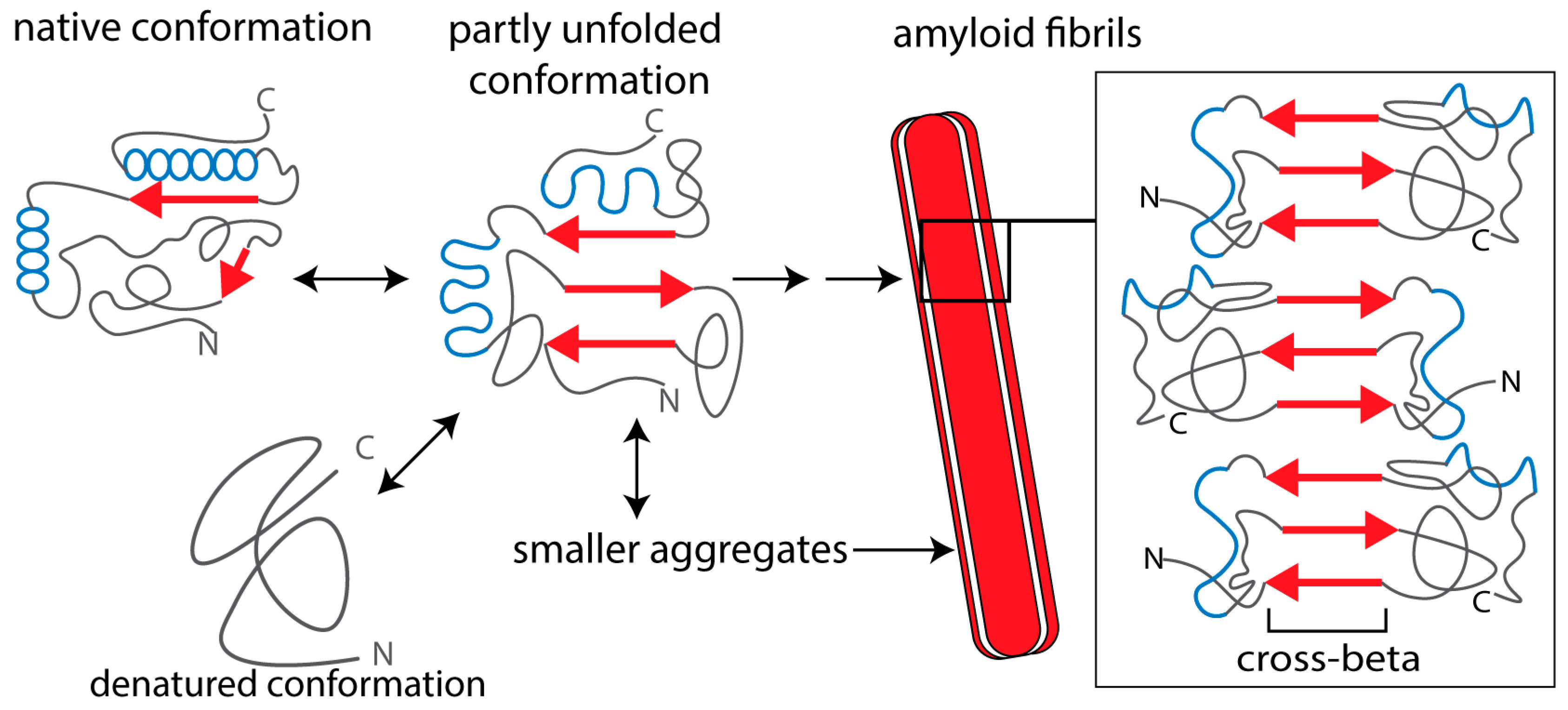
Amyloid is characterized by fibrils with diameters ranging 7–10 nm that are composed of stacked β-strands held together by hydrogen bonds [8,9,12]. Since the longitudinal axis of the amyloid fibrils is perpendicular to the plane of the β-strands, this is also referred to as the cross-β structure of amyloid fibrils (Figure 1) [8,10,12,13]. Amyloid deposits consist for ~90% of a fibril-forming protein, which is specific for each amyloid disease. However, all amyloid deposits contain common components that are involved in the amyloid formation process, such as serum amyloid P protein (SAP), heparan sulfate proteoglycans (HSPG), and apolipoprotein E (ApoE) [14].
Amyloid fibrils are present extracellularly and deposit locally at specific sites in the body, often the site of amyloid protein production, or systemically with involvement of multiple organs. Amyloid depositions cause cellular damage, tissue injury, and organ failure. For example, amyloid deposits impair appropriate supply of nutrition and oxygen to tissues or induce inflammatory reactions that cause tissue damage, including damage to nerves [15]. Based on these findings, the dogma was that mainly mature amyloid fibrils are responsible for cell dysfunction and cell death in amyloid diseases. However, fibril-forming proteins also form smaller aggregates, protofibrils, and oligomers (Figure 1), which are actually the main cause of the cellular toxicity of amyloid-forming proteins. Hence, these oligomers are often referred to as “toxic oligomers” [16].
Amyloid deposition may lead to neuropathy, as is well-established for some rare, familial amyloid neuropathies and acquired amyloid polyneuropathies [17,18,19,20,21], as well as several familial autoinflammatory syndromes [22,23]. However, for more common acquired amyloid diseases, not much is known about the development of peripheral neuropathy in these diseases. The most common disease associated with amyloid formation and peripheral neuropathy is diabetes mellitus type 2 (T2DM). Amyloid in the pancreatic islets of Langerhans is a characteristic feature of T2DM [24]. Diabetic peripheral neuropathy (DPN) is one of the most common complications of T2DM and affects nearly 50% of adults with DM during their lifetime [25]. As discussed in this review, several findings point to the amyloid protein in T2DM, islet amyloid polypeptide (IAPP), as a cause in the development of DPN. In addition, in various chronic inflammatory diseases, notably rheumatoid arthritis (RA), osteoarthritis (OA), psoriatic arthritis (PA) and inflammatory bowel diseases (IBD), both peripheral neuropathy and amyloid have been described; however, a causal relation has not yet been established.
Some indications, mainly from animal models, suggest that peripheral neuropathy may also develop in central amyloid neuropathies (notably Alzheimer’s disease, Parkinson’s disease, and prion diseases) where amyloid proteins and oligomers are also present outside the central nervous system [26,27]. However, due to their predominant central neuropathic nature, these neurodegenerative diseases will not be discussed here.
In this review, we will discuss how amyloid can cause neuropathy and address whether a common pathogenic pathway of “peripheral amyloid neuropathy” exists in both rare, familial amyloid neuropathies and acquired amyloid neuropathies, as well as in common diseases in which peripheral neuropathy and amyloid co-occur, such as T2DM and chronic inflammatory diseases, which are not (yet) considered as amyloid neuropathies.
2. Causes of Amyloidogenesis
At present, 36 proteins have been identified as amyloid fibril proteins in humans. These amyloid fibril proteins affect different tissues and are associated with diseases with highly variable prevalence [8]. The mechanisms causing aggregation or fibrillogenesis of these amyloid proteins are diverse. Various amyloidoses are characterized by the development of peripheral neuropathy. Thus, neuropathy develops in these diseases independent of the nature of the respective amyloid fibril protein and of the location of amyloid protein production [10,28] (See Table 1).
Some amyloidogenic proteins have an inherent tendency to form amyloid fibrils due to their three-dimensional structure. Among these is transthyretin (TTR), which causes wild-type TTR amyloidosis (wtATTR; also called senile systemic amyloidosis, SSA) [29] The development of amyloid in this disease is likely promoted by age-related, post-translational modifications of the TTR protein [30].
A well-established factor that promotes amyloid formation is high protein concentration. In dialysis-related amyloidosis, β2-microglobulin (β2M) concentrations in blood are elevated more than 60-fold in end-stage kidney failure due to ineffective renal clearance [31]. Similarly, serum amyloid A protein (SAA) is produced at high quantities under inflammatory conditions [32,33]. These high blood levels of SAA are essential for amyloidosis to occur; however, specific SAA gene polymorphisms are also a risk factor [23]. The notion that high protein concentration is a risk factor for amyloidosis is further supported by the finding that plasma cell or B lymphocyte proliferative disorders may cause excessive production of immunoglobulin light chains, leading to AL amyloidosis (primary systemic amyloidosis) [34]. In T2DM, IAPP is overproduced together with insulin in pancreatic islet β cells in order to compensate for insulin resistance, which promotes formation of islet amyloid deposits [24,35,36,37].
Another risk factor for amyloidosis is mutation of a gene for which the non-mutated version encodes a protein that is non-fibrillogenic. Such mutations are at the root of several hereditary forms of amyloidosis [38,39]. The causative mutations either modify proteolytic cleavage of a precursor protein or alter the protein structure, resulting in proteins with intrinsic tendency to form amyloid fibrils. In familial amyloid polyneuropathies (FAP), which are associated with peripheral neuropathy, mutations in the genes encoding TTR, gelsolin, or apolipoprotein A1 cause amyloid formation [39,40]. Amyloidosis (from the SAA protein) and peripheral neuropathy also occur in some familial autoinflammatory diseases, although the primary genetic defect in these syndromes does not occur in an amyloid protein gene [22].
3. Peripheral Amyloid Neuropathies
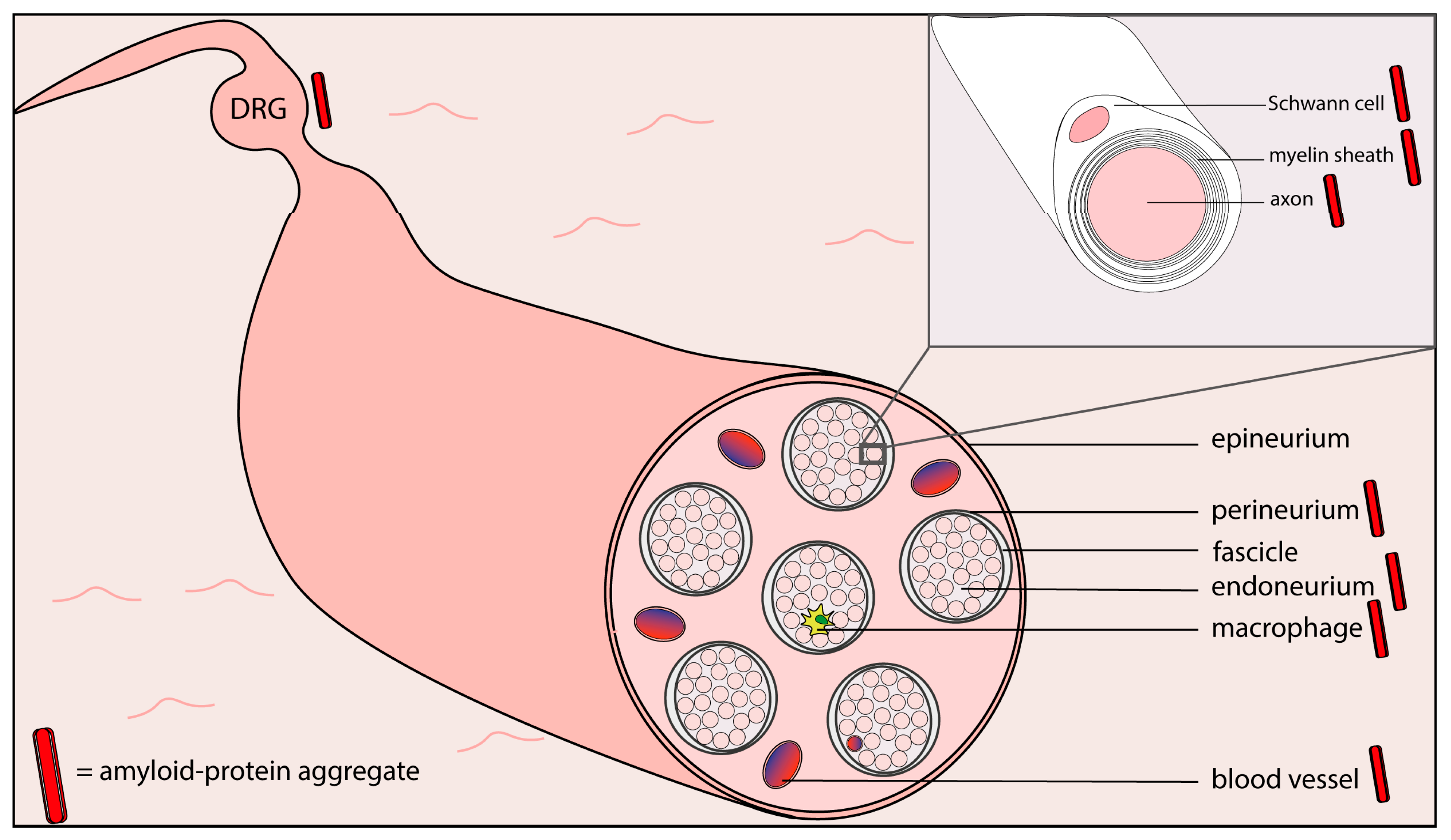
In Table 1, several characteristics of hereditary and acquired diseases with both amyloidosis and peripheral neuropathy are listed. In several of these diseases, amyloid deposits, or amyloid protein aggregates, have been found at different locations within peripheral nerves (see Figure 2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Diseases with known peripheral nervous system involvement and a (potential) link to amyloid proteins.
Table 1.
Diseases with known peripheral nervous system involvement and a (potential) link to amyloid proteins.
| Disease | Amyloid Protein | Acquired/ Hereditary | Local/ Systemic | Peripheral Nervous System Involvement | Prevalence/Incidence Disease | Prevalence Disease w/w * | Prevalence/Incidence PN (% of Patients) |
|---|---|---|---|---|---|---|---|
| Familial amyloid polyneuropathy | Transthyretin (hATTR) | Hereditary | Systemic | Polyneuropathy Autonomic disturbances Carpal tunnel syndrome [41] | 10,186 persons w/w (range: 5526–38,468)/UN [42] | 0.00013% | UN (it develops in the majority of patients/UN [42,43] |
| Apolipoprotein A-I (AApoAI) | Hereditary | Systemic | Polyneuropathy [44] | UN/UN | UN | UN/UN | |
| Gelsolin (HGA) | Hereditary | Systemic | Cranial neuropathy Polyneuropathy [28] | 400 to 1000 gene carriers in Finland/UN [45] | 0.01% | UN/UN | |
| Immunoglobulin light-chain amyloidosis | Ig light-chain | Acquired | Systemic | Polyneuropathy Autonomic disturbances Carpal tunnel syndrome [46,47] | 40.5 cases per million in the US/9.7 to 14.0 cases per million per year in US [48] | 0.004% | 15–20%/UN [49] |
| Dialysis-related amyloidosis | β2-microglobulin | Acquired | Systemic | Carpal tunnel syndrome Polyneuropathy [50,51] | UN/UN (incidence > 95% in. patients > 15 years dialysis in US) [51] | UN | UN/UN |
| Senile systemic amyloidosis | Transthyretin | Acquired | Systemic | Polyneuropathy Autonomic disturbances carpal tunnel syndrome [52] | 63/256 of the study population in Finland (25% > 80 years old)/UN [53] | 0.45% | UN/UN |
| Type 2 diabetes mellitus | IAPP | Acquired | Local/systemic | Polyneuropathy [54] | 463 million persons (aged 20–79 years) w/w (including T1DM&T2DM)/UN [55] | 5.4% | 31.5–45% [56,57] |
| Rheumatoid arthritis | SAA | Acquired | Systemic | Polyneuropathy [58] | 19,965,115 persons w/w/1,204,599 new cases w/w [59] | 0.26% | 39.19–75.28% [58] |
| Inflammatory bowel disease | SAA | Acquired | Systemic | Polyneuropathy [60,61] | 68 million persons w/w/70,000 new cases per year in USA [62,63] | 0.09% | UN/0.07% after 10 years of IBD [60,64] |
| Osteoarthritis | TTR, Apo-A1 | Acquired | Systemic | Polyneuropathy [65] | 303 million persons w/w (80% of people > 75 years)/14.93 million new cases w/w [66,67] | 3.9% | UN/UN |
| Psoriatic Arthritis | SAA | Acquired | Systemic | Polyneuropathy [68] | 133 per 100,000 persons w/w/83 per 100,000 persons per year w/w [69] | 0.133% | UN/UN |
| Familial Mediterranean fever | SAA | Hereditary | Systemic | Polyneuropathy [22] | 100,000 persons in Turkey /UN (high among people from the eastern Mediterranean e.g., Arabs, Turks, Jews, and Armenians) [70,71] | 0.13% | UN/UN |
| Muckle–Wells syndrome | SAA | Hereditary | Systemic | Polyneuropathy [22] | Rare, MWS is one of the three clinical forms of CAPS and the prevalence of CAPS is 1–10 cases per million in France/UN [72] | 0.001% (based on max 10 per million) | UN/UN |
Abbreviations: CAPS = cryopyrin-associated periodic syndrome; HGA = hereditary gelsolin amyloidosis; hATTR = transthyretin-associated hereditary amyloidosis; IAPP = islet amyloid polypeptide; IGT = impaired glucose tolerance; MWS = Muckle–Wells syndrome; PN = peripheral neuropathy; SAA = serum amyloid A protein; T1DM = type 1 diabetes mellitus; T2DM = type 2 diabetes mellitus, UN = unknown; w/w = worldwide, * The calculated numbers for estimation of the worldwide prevalence are based on the: (1) same prevalence in every country; (2) world population = 7.6 billion, worldwide population aged over 80 years = 143 million, US population = 327 million, Finnish population = 5.5 million, Turkish population = 80 million. Although the prevalence of some diseases may be very different in different parts of the world, these (albeit artificial or fictional) numbers enable a global comparison of the prevalence of these diseases.
3.1. Familial and Acquired Amyloid Polyneuropathies
Familial amyloid polyneuropathy (FAP) is the collective name for three inherited autosomal dominant disorders caused by germline mutations in genes encoding amyloidogenic proteins. Acquired neuropathic amyloidosis refers to systemic amyloidosis, which are associated with peripheral neuropathy but are not caused by a mutation. These diseases may develop secondary to another disease or as a consequence of aging. Among 36 amyloid fibril proteins identified in humans, only a few have been associated with acquired peripheral neuropathy [8,82].
Although the prevalence of peripheral neuropathy in the various amyloid diseases may be quite variable (see [83]) and the relevance of amyloid protein aggregation for the development of peripheral neuropathy in these disease may also vary, some common characteristics do emerge. Peripheral amyloid neuropathy usually presents as symmetric polyneuropathy, which is length-dependent, i.e., beginning in the lower extremities and extending more proximally as the disease progresses [84,85,86]. This nature of the disease, primarily affecting the longest axons, is likely due to defects in the axonal transport [87] or mitochondrial function [79,80,81,88], because both are important for maintaining the integrity of long axons. Initially, both myelinated and unmyelinated small fibers are affected, with decreasing nerve fiber density in later stages of the disease [89]. With disease progression, large-myelinated fibers are also affected and loss of motor neurons even occurs in some of these diseases [40]. In addition to peripheral sensory nerves and DRGs [74,90,91], amyloid has also been detected in autonomic nerves [92] and autonomic ganglia [21]. As a consequence, autonomic dysfunction is present in several of the amyloid diseases (Table 1 and [83]). Development of signs and symptoms of autonomic dysfunction, however, usually occurs later than the peripheral polyneuropathy [93].
Amyloid and oligomers have been detected several years before the beginning of neuropathy [75,94,95,96], suggesting that amyloid protein aggregation may be involved early in the pathogenesis of the neuropathy. For hereditary apolipoprotein A-I amyloidosis (AApoAI), various ApoA-I gene mutants cause (poly) neuropathy, however not all amyloidogenic apoA-I gene mutations lead to neuropathy [97], indicating a genotype–phenotype correlation. Neuropathy in hereditary gelsolin amyloidosis (HGA) might be caused by both amyloid formation and loss of function of the gelsolin protein [98], since gelsolin is a key regulator of actin filament assembly and disassembly [99,100], and in neurons these processes govern spine formation, morphology, and synaptic functions [101]. For the familial autoinflammatory syndromes, familial Mediterranean fever and Muckle–Wells syndrome, amyloidosis from the SAA protein is suggested as cause of the peripheral neuropathy, although the primary genetic defect occurs in another gene [22,23].
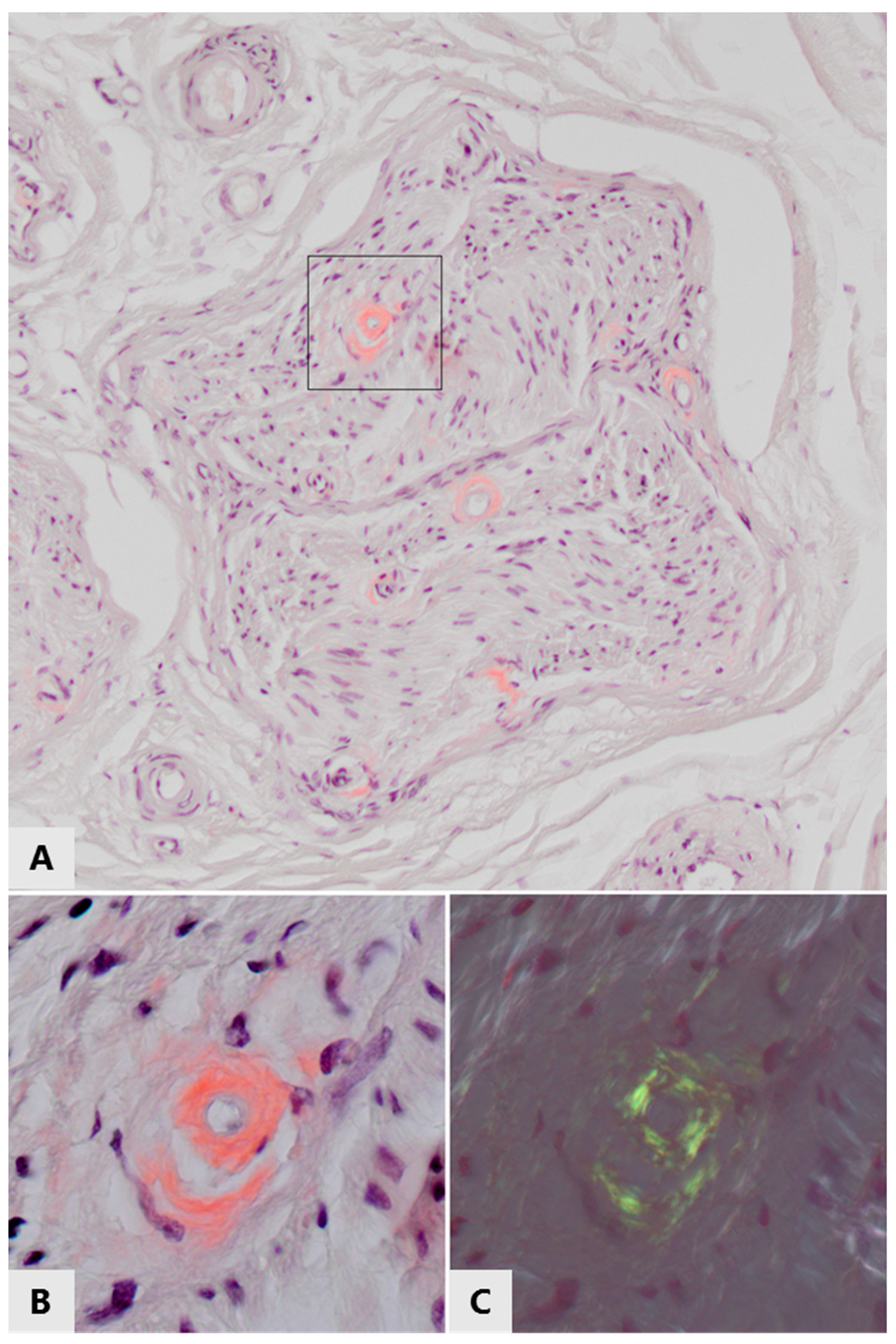
AL amyloidosis, an acquired neuropathic amyloidosis, is caused by overproduction of antibodies in plasma cell or B lymphocyte proliferative disorders, such as multiple myeloma. Immunoglobulin light chains aggregate and form amyloid deposits, for example in nerves ([48,102,103,104,105], see Figure 3). In dialysis-related amyloidosis (DRA), β2 microglobulin gradually accumulates in blood when it cannot pass the dialysis membrane and the most frequent polyneuropathy is distal axonal sensorimotor polyneuropathy [106,107,108]. Thanks to general improvements in dialysis care, the prevalence of this type of amyloidosis appears to be decreasing [109]. In senile systemic amyloidosis (SSA), age-related deposition of wtTTR amyloid is found in several organs, while carpal tunnel syndrome is often the first clinical manifestation of the disease [29,110]. Oxidation of methionine and cysteine residues of TTR increases with age and promotes aggregation of wild-type TTR; therefore, these post-translational modifications have been implicated in the pathogenesis of SSA [30]. In contrast to hATTRv, a subtype of FAP where neuropathy is frequent, neuropathy is either not frequent or underdiagnosed in SSA [28], suggesting that mutant TTR is more neurotoxic than wtTTR.
Both in familial amyloid polyneuropathies and in acquired amyloid polyneuropathies, other organs than the nervous system can be affected, notably the liver, kidney, heart, and gastrointestinal tract [44,53,92,111,112,113,114,115,116,117]. Why these particular tissues are commonly affected is not known.
3.2. Common Acquired Diseases with Peripheral Neuropathy and Amyloid
Apart from the mostly rare amyloid neuropathies described above, some common acquired diseases also are associated with peripheral neuropathy and amyloid. However, little is known about the potential contribution of the amyloidogenic protein in the pathogenesis of peripheral neuropathy in these diseases, and these diseases are not (yet) classified as peripheral amyloid neuropathies. The possible causative relation of amyloid and toxic oligomers with peripheral neuropathy in these diseases will be discussed next, with a specific focus on type 2 diabetes mellitus.
3.2.1. Type 2 Diabetes Mellitus (T2DM)
In 2019 the International Diabetes Federation (IDF) indicated that there were ~450 million people with DM worldwide. This number is expected to rise to 700 million by 2045 [55]. Type 1 DM is characterized by autoimmune-mediated loss of the insulin-producing β-cells in the pancreatic islets of Langerhans, causing insulin insufficiency and hyperglycemia [118]. T2DM is the most common type of DM, accounting for approximately 90% of all DM patients [55]. T2DM is characterized by both insulin resistance (reduced insulin sensitivity of insulin target tissues as a consequence of obesity) and β-cell failure (insulin insufficiency), leading to hyperglycemia. Increased β-cell apoptosis in T2DM is associated with glucotoxicity, lipotoxicity, and deposition of amyloid in the pancreatic islets [119,120,121]. Islet amyloid is a characteristic histopathological feature of T2DM, being detected in approximately 90% of T2DM patients at autopsy [24]. However, islet amyloid has recently also been detected in 3 young patients with T1DM [122].
Peripheral neuropathy is the most frequent chronic complication of DM. The prevalence of peripheral neuropathy in DM ranges from 10% at one year after DM diagnosis to more than 50% during progression of the disease [4,25,123,124], making diabetic peripheral neuropathy (DPN) the most abundant type of peripheral neuropathy worldwide [3,4,125] (Table 1). Diabetic neuropathy that is painful develops in approximately 50% of DM patients with neuropathy [126]. DPN is a major cause of lower limb amputation, which severely affects both quality of life and life expectancy [127]. Peripheral neuropathy in T2DM is poorly managed clinically because of its late diagnosis, complex pathogenesis, and the limited therapeutic options to treat neuropathy [4]. Long-lasting DM causes loss of sensory peripheral nerve terminals. At the early stage of DPN, small nociceptive sensory fibers are commonly affected. Motor function is hardly affected, although some slowing of motor conduction velocity is observed [128]. The symptoms of DPN involve gain- or loss-of-function, depending on the type of nerve that has been damaged. Gain-of-function symptoms include allodynia (feeling of pain from non-painful stimuli) and hyperalgesia (increased pain sensitivity), whereas loss-of-function symptoms include tactile and thermal hyposensitivity [3,4]. Patients can experience pain in some areas of the body and loss of sensitivity in other areas [3,124]. During progression of DPN, patients can even develop a total loss of sensation (numbness), which contributes to development of complications such as diabetic foot ulcers [25]. The symptoms of DPN tend to follow a “stockings and gloves” pattern, which means that they start at the feet and hands [3,124].
Hyperglycemia is generally considered a primary cause of DPN [3,124]. Several hyperglycemia-induced molecular pathways contribute to deregulation of neuronal function, including the polyol pathway, hexosamine pathway, activation of PKC isoforms (notably α, β1, β2, δ, and ε), and formation of advanced glycation end products (AGEs), among others [3,129,130,131,132,133]. These pathways and molecules cause microangiopathy, oxidative stress, and inflammation, which contribute to cytotoxic effects on neurons and Schwann cells, leading to nerve fiber loss and axonal degeneration, and consequently loss of sensory perception (reviewed in [130,131,132,133,134]). However, some data indicate that other factors besides hyperglycemia play a role in the development of DPN in T2DM. For example, tight blood glucose control is able to reduce hyperglycemia and diminish neuropathy in T1DM [135], but in T2DM improved glycaemia is not, or only partly, accompanied by less severe neuropathy [135,136]. Moreover, neuropathy is also present in individuals with prediabetes (i.e., not yet having developed hyperglycemia) [137], indicating factors other than hyperglycemia are involved. Large clinical studies support the concept that components of the metabolic syndrome, (notably obesity and prediabetes) which include elevated levels of the amyloidogenic protein hIAPP, may underlie the pathogenesis of DPN, especially in T2DM [138].
IAPP is the fibril-forming protein of pancreatic islet amyloid, which is composed of 37 amino acids and is co-produced and co-secreted with insulin from the pancreatic islet β-cells [36,139,140]. The physiological functions of IAPP are not fully understood, but include enhancement of satiety, reduction of gastric emptying and of glucagon release, and inhibition of insulin signaling [36,139]. Insulin resistance leads to increased production of insulin to compensate for its impaired signaling, which is accompanied by increased production of IAPP [35,36]. Moreover, free fatty acid levels in the blood increase with obesity, promoting IAPP gene expression and secretion by islet β-cells [141]. Elevated concentrations of human IAPP (hIAPP) trigger formation of toxic oligomers and amyloid plaques, which impair islet function and increase β-cell apoptosis [24,140,142]. In contrast to hIAPP and IAPP from monkeys and cats, murine and rat IAPP are not amyloidogenic due to differences in the amino acid sequence [143,144]. Therefore, rodent models with islet β-cell-specific expression of a hIAPP transgene have been developed to study the pathogenic role of hIAPP and islet amyloidosis [145]. In T2DM, deposits of aggregated hIAPP are found in the pancreatic islets, but also elsewhere in the body, i.e., in the heart, kidneys, and brain [146,147,148], indicating that amyloidosis in T2DM is not restricted to the pancreas. Apart from the pancreatic islet β-cells, IAPP is also expressed in peptidergic sensory neurons [149]. Several mouse studies revealed that IAPP has an excitatory role in nociception [150,151,152,153,154,155]. Therefore, with the knowledge that several other amyloid proteins cause peripheral neuropathy, we hypothesized that aggregated hIAPP causes peripheral neuropathy in individuals where hIAPP is overproduced, as is the case in (development of) T2DM.
Injection of hIAPP in wild-type mice induces mechanical hypersensitivity and reduction in nerve fiber density [156]. More importantly, in a more physiologically relevant model system, i.e., transgenic mice endogenously expressing hIAPP specifically in pancreatic islet β cells [142,157], mechanical hypersensitivity develops and skin nerve fibers are reduced [156]. Thus, hIAPP causes signs of peripheral neuropathy in vivo, even in the absence of hyperglycemia. In further support of hIAPP as a driver of painful neuropathy, others reported that IAPP modulates neuropathic pain in mice and rats at different levels of the nervous system [154,158]. Moreover, hIAPP is involved in central neuropathy, both in Alzheimer’s disease patients with T2DM [159] and in diabetic hIAPP transgenic rats [159,160], further substantiating the neuropathic potential of aggregated hIAPP. Overall, these data support the hypothesis that hIAPP is a driver of peripheral neuropathy in T2DM, but since the clinical data are only correlative, further investigation into the role of hIAPP in peripheral neuropathy in human T2DM is warranted.
3.2.2. Acquired Chronic Inflammatory Diseases
Serum amyloid A protein amyloidosis (SAA) is a major complication of chronic inflammation and one of the most common human systemic amyloid diseases worldwide. Serum amyloid A protein is synthetized in large quantities in chronic inflammatory diseases and can lead to amyloid deposits in any chronic inflammatory disorder [161,162]. Amyloid deposits are mainly found in kidney, subcutaneous adipose tissue, and gastrointestinal mucosa. Peripheral neuropathy is not considered as a characteristic feature of this systemic amyloid disease. However, in several chronic inflammatory diseases, notably in rheumatoid arthritis, osteoarthritis, psoriatic arthritis, and to a lesser extent inflammatory bowel diseases, peripheral neuropathy is present in a subset of patients, and amyloid has been demonstrated in these diseases [58,163] (see Table 1). For example, in sural nerve biopsies of rheumatoid arthritis patients, perineurial thickening with amyloid was detected in 4 out of 23 patients investigated [163]. In knee joints of 12 out of 12 osteoarthritis patients, as well as 7 out of 12 aged individuals without osteoarthritis, amyloid was present in menisci, articular cartilage, and synovial membranes, mostly of TTR and Apo-AI origin [164,165]. In psoriatic arthritis, amyloid has been reported in 30 cases and was present mainly in the kidney [166]. In Crohn’s disease, SAA-protein-derived amyloid is found in the kidney and gastrointestinal tract in 0.3–10% of patients [167,168,169].
Whether there is a causal relation between the development of peripheral neuropathy and amyloid in these diseases is not known, because literature on peripheral nervous system involvement in inflammation-related amyloidosis is scarce. However, three cases reports of patients with peripheral neuropathy and SAA protein amyloid exclusively within axons and myelin sheaths underscore a potential link between SAA amyloid and peripheral neuropathy [78]. Thus, amyloid protein aggregation might also be involved in development of peripheral neuropathy in chronic inflammatory diseases.
The presence of amyloid in histological specimens is routinely demonstrated by Congo red staining of tissue sections and subsequent yellow-green birefringence when viewed with polarized light (see Figure 3). The reported prevalence of amyloid detected with this method within the peripheral nervous system in T2DM and chronic inflammatory diseases is low, or has even not been investigated or reported at all. However, this technique only detects fibrillary amyloid deposits, not prefibrillar aggregates such as oligomers. Notably, these oligomers are generally thought to be the most cytotoxic species of amyloid protein aggregates. In addition, amyloid deposits are generally larger, and thus more readily detectable in organs such as the kidney and heart as compared to nerves or even individual neurons. We propose that pathogenic involvement of amyloid protein aggregation in peripheral neuropathy may be underestimated in some diseases not (yet) considered as amyloid neuropathies.
As peripheral sensory neuropathy is a hallmark of several amyloidoses, T2DM, and some inflammatory diseases, the question arises as to why sensory neuropathy is most prominent in these diseases. Damage to peripheral sensory nerves may be noticed or diagnosed sooner as compared to other nerves. Nevertheless, peripheral neurons, in particular long axons, may be particularly sensitive to degeneration and to toxic compounds due to their effects on key processes essential to maintaining homeostasis in these long axons, such as protein transport, membrane integrity, and mitochondrial function. Some sensory neurons are not covered by myelin sheets, which may render them even more susceptible. Finally, sensory neurons may have a particular sensitivity towards oligomers because of the composition of plasma membrane lipids, which might promote oligomer toxicity. Although these aspects may contribute to the observed presence of neuropathy in amyloidosis, future research will have to address these issues.
To further support potential links between amyloid or toxic oligomers and the development of peripheral neuropathy in these diseases, we will discuss the mechanisms as to how amyloid and amyloid protein aggregates may contribute to peripheral neuropathy.
4. Mechanisms Linking Amyloid and Peripheral Neuropathy
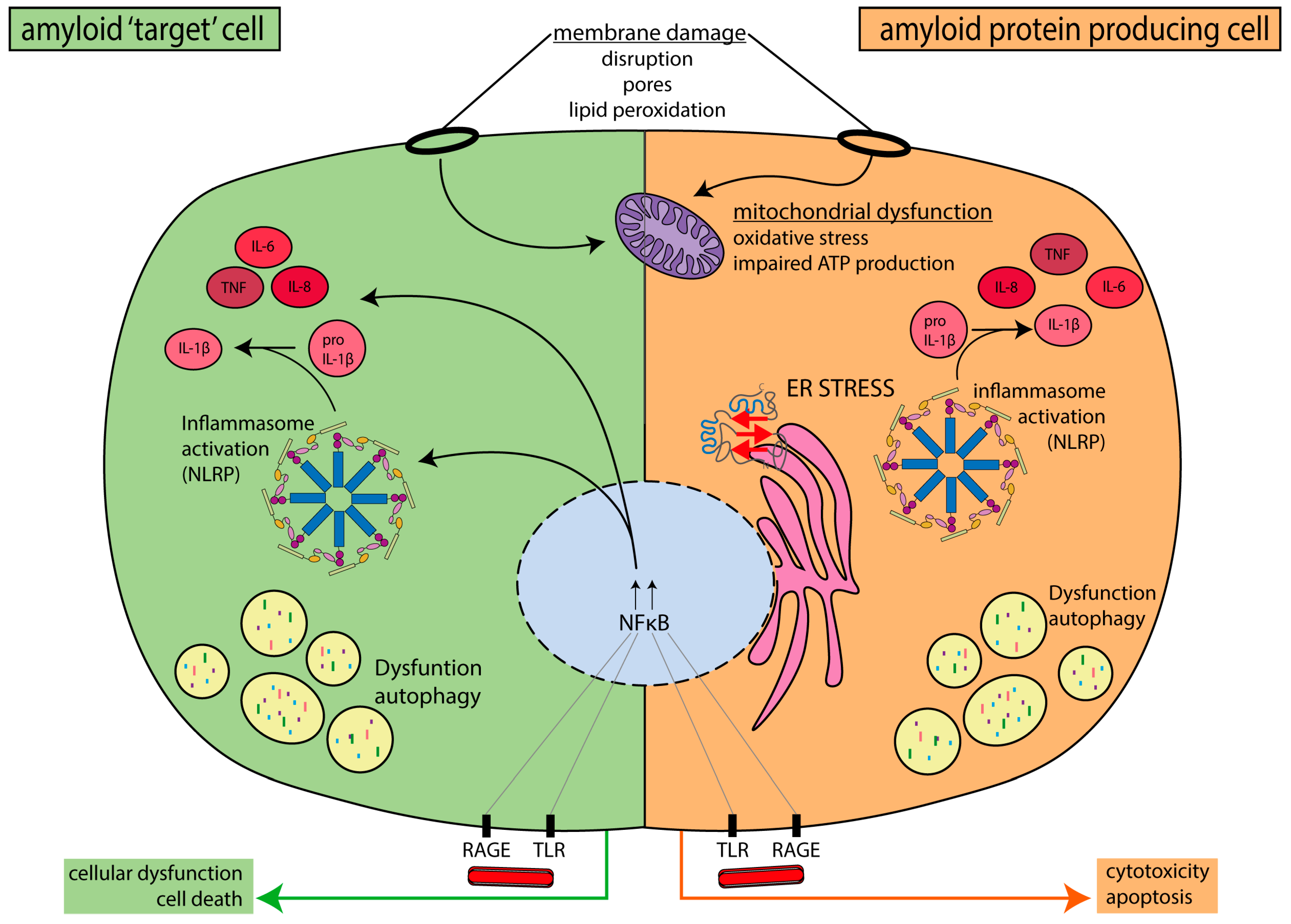
Amyloid and toxic oligomers may cause cell damage through various mechanisms, including membrane disruption, impaired mitochondrial function, autophagy dysfunction, and others. Many of these cellular deficits are involved in the development of peripheral neuropathy. These deficits may occur not only in amyloid-protein-producing cell types, such as hepatocytes, pancreatic islet β-cells, and neurons, but also in other cell types involved in the development of neuropathy, such as macrophages or microglia, Schwann cells, and endothelial cells. The information described below on the cell biological mechanisms in the pathogenesis of amyloid toxicity, including (peripheral) neuropathy, is summarized in Figure 4.
4.1. Protein–Membrane Interactions
Interaction of amyloid proteins with membrane phospholipids promotes fibril formation of SAA [170,171,172], Aβ [173,174], hIAPP [175,176], and apolipoprotein C-II [177]. In vitro, hIAPP amyloid fibril formation occurs at or in the cell membrane [175]. When interacting with membranes, amyloid proteins may adopt α-helical conformations that further promote aggregation [178,179]. Membranes are also a target for cytotoxic actions of amyloid protein aggregates, as is extensively demonstrated for SAA [180], hIAPP, Aβ, and α-synuclein [181,182,183]. Amyloid proteins disrupt the integrity of the phospholipid bilayer, but oligomers also form small pores in the plasma membrane that can act as non-specific ion channels, called “amyloid pores” [184,185]. In neurons, such pores could lead to membrane depolarization, causing ectopic discharges and neuronal damage. Basal membranes and organelle membranes (e.g., of the mitochondria, endoplasmic reticulum, and nucleus) are also affected by amyloid protein aggregates [186,187,188]. Amyloid-mediated disruption of membrane integrity increases the exposure of polyunsaturated fatty acids to cytosolic reactive oxygen species (ROS) leading to formation of reactive aldehydes. For example, Aβ causes hydroxyl radical generation, which induces membrane lipid peroxidation through hydrogen abstraction from polyunsaturated fatty acids [189]. Generated reactive aldehydes further elevate ROS production and trigger inflammatory responses [190]. Human IAPP and Aβ cause membrane lipid peroxidation in primary neurons, which triggers calcium influx and IL-lβ synthesis [159,191]. Inhibitors of lipid peroxidation reduced mechanical hypersensitivity in a rat model of neuropathic pain [192], supporting a role of lipid peroxidation in the development of peripheral neuropathy.
4.2. Endoplasmic Reticulum (ER) Stress
The endoplasmic reticulum (ER) is the main cellular compartment involved in protein folding and secretion. Disturbances in ER homeostasis (e.g., due to aberrant or misfolded proteins) induce ER stress. ER stress triggers the activation of the unfolded protein response (UPR), a response which alters the expression of genes involved in ER quality control. The UPR aims to recover ER homeostasis or trigger apoptosis when cells are irreversibly damaged. As cells age, their ability to maintain a balance in protein folding or degradation and to cope with disruptions in proteostasis declines. This decline contributes to the development of age-related diseases, including neurodegenerative diseases [193,194] and putatively acquired amyloidosis, along with its complications.
Amyloidogenic proteins, either wild-type or mutant, have a tendency to misfold and aggregate, which puts pressure on the ER, particularly in cases of overproduction (e.g., SAA in inflammation and hIAPP in insulin resistance and T2DM) or production of a mutant protein with acquired amyloidogenic potential. Thus, it is not surprising that ER stress is a major mechanism implicated in cytotoxicity in amyloidoses. Increasing evidence indicates that the toxic molecular species that evoke ER stress are the intermediate oligomeric forms and not the mature amyloid fibrils [195].
In systemic amyloidoses, the amyloidogenic proteins evade ER quality control, causing progressive aggregation and amyloid deposition. Regulation of ER quality control, therefore, is a crucial mechanism in defining the onset and progression of systemic amyloid diseases [196]. TTR aggregates and ER stress are present in dorsal root ganglia of TTR transgenic mice [197], suggesting a role in familial amyloid polyneuropathy.
For hIAPP and Aβ, there is extensive evidence that these proteins cause ER stress in cell and mouse models of T2DM [198,199,200,201,202,203,204] and AD [205,206,207], respectively. ER stress is also a determining factor for the onset and progression of diabetic peripheral neuropathy (DPN) [208,209]. ER stress is involved in DPN in rodent models of T2DM [209]. For example, in diabetic rats, sensory neurons display ER stress, and inhibitors of ER stress reduce the development of neuropathic pain in these animals [210]. Moreover, reduction in ER stress with a chemical chaperone in diabetic rats reduces DPN, despite continuous hyperglycemia. Thus, ER stress independent of hyperglycemia promotes peripheral nerve damage [211]. Conversely, induction of ER stress in healthy animals generates an immediate and lasting painful phenotype that is reversible by ER stress blockers [210]. Finally, ER stress is involved in other forms of peripheral neuropathy (see [212]), pointing to a central role of ER stress in peripheral neuropathy in general.
4.3. Mitochondrial Dysfunction
Neurons are cells with a complex morphology, long lifespan, and high energetic requirements. Mitochondria are essential energy suppliers that maintain the viability and proper function of neurons. Mitochondria generate ATP, buffer cytoplasmic calcium (Ca2+) levels, and are a source of metabolites required for neuronal functioning [213,214,215]. Thus, neurons are highly dependent on adequate mitochondrial function to maintain neuronal integrity and activity [79,80,81]. Because of the unique length and energy requirements of peripheral nerves, appropriate mitochondrial function and distribution along nerves is of fundamental importance. In general, mitochondrial dysfunction, which may lead to oxidative stress, is a main cause of axonal injury in peripheral neuropathy. Mutations in genes involved in mitochondrial fusion, fission, and axonal transport are linked to inherited peripheral nerve diseases, with demyelination and axonal defects as a consequence [88]. Moreover, peripheral neuropathy is associated with mitochondrial dysfunction, such as reduced oxidative phosphorylation, reduced ATP production, increased production of reactive oxygen species (ROS), and altered mitochondrial transport (reviewed in [216,217]).
For amyloid neuropathy, some genetic evidence supports mitochondrial involvement. Polymorphisms in mitochondrial genes are associated with early onset of disease, i.e., with a progressive sensorimotor and autonomic axonal polyneuropathy [218], and with a sex-specific effect on penetrance of the disease in FAP patients or families [219].
Although experimental proof for mitochondrial dysfunction in peripheral amyloid neuropathy is lacking, overwhelming evidence exists that impaired mitochondrial function is a causative factor in T2DM (reviewed in [220]) and in the central amyloid neuropathies, such as AD and Parkinson’s disease [187,221,222,223]. Common cellular defects caused by hIAPP and Aβ include reduced activity of mitochondrial respiration and increased ROS generation [224]. Accumulation of hIAPP oligomers on mitochondrial membranes causes mitochondrial damage and β-cell toxicity, whereas an IAPP-specific ligand that binds IAPP and rescues β-cells from hIAPP-induced cytotoxicity [179] prevents mitochondrial membrane pores and toxicity [225]. Aβ oligomers downregulate mitochondrial oxidative phosphorylation [226], decrease mitochondrial potential, and increase ROS generation in rat neurons [227]. Oligomers of α-synuclein reduce axonal mitochondrial transport, disrupt axonal integrity, and reduce ATP levels in human neurons. These defects were restored by inhibition of oligomer formation [228].
Thus, oligomers of amyloidogenic proteins cause mitochondrial defects that play an important role in T2DM and in several neurodegenerative diseases, and likely also in amyloid-induced peripheral neuropathies.
4.4. Inflammation
Inflammation is a consequence and a cause of amyloidosis and amyloid neuropathy. In hereditary amyloidosis, inflammation results from the amyloidosis and contributes to development of the associated pathology, whereas in SAA amyloidosis, chronic inflammatory diseases are the cause for amyloid formation.
For familial amyloid neuropathies, most evidence that amyloid triggers inflammatory processes and neuropathy comes from hTTRv. In FAP/TTR, plasma levels of prototypic inflammatory cytokines are increased. Interestingly, IL-1β, IL-10, and IL-33 levels were increased already before any presence of amyloid fibril deposition in one study [229], indicating that prefibrillar aggregates caused the increase in inflammatory mediators. Early in FAP development, non-fibrillar TTR aggregates are detected in nerve biopsies that are associated with signs of inflammation, e.g., increased expression of the proinflammatory cytokines TNF and IL-1β and iNOS in nerves [90]. Toxic protein aggregates cause inflammation through activating the receptor for advanced glycation end products (RAGE) and Toll-like receptors (TLR) [230,231], which activate the transcription factor NFκB, resulting in proinflammatory cytokine gene expression [232,233]. For example, in vitro, TTR binds RAGE and activates NFκB in transfected PC-12 cells [234]. TTR fibrils also induce cytokine and iNOS expression by Schwann cells and endothelial cells in vitro in a RAGE-dependent manner [75]. Evidence implicates RAGE signalling in the pathogenesis of neuropathic pain. RAGE causes Wallerian degeneration via modulation of the inflammatory response [235], and causes sensory neuron damage in vitro by the activation of NF-κB, JAK-STAT, and ERK pathways [236]. Moreover, pharmacological inhibition of RAGE effectively attenuates development of chronic inflammatory and neuropathic pain [237]. In general, in models of neuropathic pain, inflammatory responses, including cytokine responses such as increased IL-1β and TNF, are thought to contribute to the neuropathic pain [238,239,240,241,242].
In the more common amyloid diseases with neuropathy, similar inflammatory reactions are observed as in FAP. The pyrin domain containing NOD-like receptor (NLRP3) inflammasome is a key component of the innate immune system that induces proinflammatory cytokine production. Protein aggregates from hIAPP and Aβ activate NLRP3 [243]. Similarly, aggregated SAA induces NLRP3 activation and IL-1β release in human neutrophils and keratinocytes of psoriasis patients with SAA amyloidosis [244,245]. SAA also triggers inflammatory responses through activation of TLR2, inducing IL-6 and IL-8 secretion in fibroblasts [246]. In T2DM and in obesity, pancreatic islet inflammation is implicated in β-cell dysfunction [247,248]. Extracellularly located hIAPP oligomers (but not fibrils or monomers) bind and activate RAGE, causing islet β-cell inflammation and apoptosis, whereas genetic or pharmacological inhibition of RAGE prevents hIAPP-induced β-cell apoptosis and islet inflammation [249]. Whether hIAPP-induced inflammation is the main cause for human DPN in T2DM is not yet known, but hIAPP clearly induces inflammation [249,250,251,252] and neuropathic pain [156]. In addition, accumulation of hIAPP in the brains of transgenic rats and injection of aggregated hIAPP in mice are linked to neuroinflammation and neurologic deficits [159]. Finally, serum TNF levels are higher in T2DM patients with peripheral neuropathy as compared to T2DM patients without peripheral neuropathy, providing further support for an inflammatory component in development of peripheral neuropathy in T2DM [253].
Additionally, local neuroinflammation is thought to be a consequence of protein aggregation in AD [254]. Aβ-induced neuroinflammation is mediated by the neuronal NLRP1 inflammasome [255], RAGE, [253,254,255,256,257] and TLR4 [256,257] in neurons. Compelling evidence has demonstrated the contribution of neuroinflammation to the pathogenesis of AD (reviewed in [258,259,260]).
Apart from amyloid-protein-producing neurons and amyloid-clearing macrophages and microglia (discussed next), other cell types in peripheral nerves can also contribute to proinflammatory cytokine production in peripheral neuropathy.
Schwann cells express RAGE and several TLRs (with upregulation of TLR-1 in injured nerves) and inflammatory cytokine expression of Schwann cells (IL-1β, IL-6 and TNF) is upregulated following peripheral nerve injury [261]. Endothelial cells express RAGE, TLRs, NLRP-1, and NLRP-3. Activation of these receptors in response to stimuli from the bloodstream, including pathogens and damage signals, can trigger production of proinflammatory cytokines, such as IL-1β, IL-6, IL-18, and TNF [262,263,264]. Thus, both endogenously produced amyloid proteins and extracellular amyloid protein aggregates promote cytokine production by several cell types, contributing to an inflammatory phenotype that promotes apoptosis in peripheral amyloid neuropathy.
4.5. Macrophages and Microglia
Macrophages are central to the induction of inflammatory responses by amyloid or protein aggregates. Amyloid strongly activates macrophages to clear protein aggregates, but also causes inflammatory responses and cell death due to the inability of macrophages to process amyloid effectively. Overall, these processes may lead to neurotoxicity. For example, macrophages in the sural nerve of AL patients are activated by amyloid and phagocytose and degrade amyloid protein aggregates and fibrils [73]. Similarly, macrophages are engaged in removal of TTR deposits in FAP patients [240] and FAP mouse models [265,266,267]. Macrophages also phagocytose β2M amyloid fibrils. However, lysosomal processing of β2M fibrils by macrophages is impaired as compared to degradation of non-fibrillary SAP protein [268]. This “frustrated phagocytosis” of amyloid fibrils may contribute to death of macrophages, because endocytosed β2M amyloid fibrils cause disruption of endosomal and lysosomal membranes [269]. As a consequence of this inability of macrophages to process amyloid deposits or amyloid fibrils effectively, tissue macrophage numbers decline, which may lead to further amyloid accumulation and consequent progression of the neuropathy. Indeed, reduced numbers of tissue macrophages as compared to non-diseased control tissue were observed in amyloid-containing hearts of FAP patients [270]. Moreover, in a mouse model expressing the V30M TTR mutant, impairing macrophage recruitment increased amyloid load and expression of apoptotic cell markers [267]. These data indicate that macrophage insufficiency may lead to amyloid accumulation and disease progression in FAP patients.
Beside causing cell death of macrophages, amyloid proteins directly activate macrophages to produce cytokines. SAA stimulates Il-1β expression in mouse and human macrophages [271,272,273]. In T2DM, macrophages serve a dual role; in early stages of hIAPP aggregation they phagocytose and degrade hIAPP aggregates and dead β-cells, while by producing IL-1β they impair islet function [252]. Mechanistically, hIAPP aggregates stimulate resident islet macrophages via TLR signalling and NLRP3 inflammasome activation to produce IL-1β, which causes β–cell apoptosis [250,251,252,274]. In support of this notion, clinical trials aimed at inhibiting IL-1β in T2DM patients showed improved β-cell function [247]. With regard to DPN, diabetes-induced neuropathy is associated with infiltration of blood-derived macrophages in the spinal cord [275], a proinflammatory phenotype of blood monocyte-derived macrophages [276] and activated spinal microglia [277].
Not surprisingly, amyloid proteins also activate microglia, the macrophages of the CNS. Microglia stimulate clearance of amyloid plaques, but amyloid-activated microglia release inflammatory cytokines, chemokines, and free radicals that may damage neurons. In vitro, a mutant form of TTR (A25T) activates microglia to secrete TNF, IL-6, and nitric oxide, and exposure of neuronal cultures to media conditioned by fibril-activated microglia causes synapse loss and ultimately extensive neuronal apoptosis, whilst A25T TTR fibrils are not directly toxic to neurons [278]. In vivo, intracerebroventricular injection of A25T TTR fibrils in mice induces microgliosis, increases brain TNF and IL-6 levels, and induces cognitive deficits that are prevented by the microglia inhibitor minocycline [278]. Other amyloid proteins also engage microglia. SAA stimulates IL-1β expression in rodent microglia [272,273,279]. Similar to macrophages, microglia activation by aggregated Aβ- and α-synuclein involves RAGE, TLR, and NLRP3 [280,281,282,283]. Importantly, nervous tissue infiltration or proliferation of resident macrophages and activated spinal microglia are key for initiation and maintenance of neuropathic pain [284,285], including painful diabetic neuropathy [286,287]. Thus, the presence of amyloid in nervous tissue could either aggravate macrophage and microglia responses or even initiate macrophage and microglia accumulation, promoting neuropathic pain in amyloid disease.
4.6. Autophagy Impairment
Autophagy is responsible for the bulk degradation of misfolded protein aggregates and damaged organelles through the lysosomal machinery. The proteasome and autophagy–lysosomal pathways are the two major routes for intracellular clearance of mutant or misfolded proteins, and protein degradation is a key cellular mechanism for protein homeostasis and cell survival. Both for cells endogenously producing amyloid proteins and macrophages and microglia removing amyloid deposits, degradation of aggregated proteins involves the autophagy–lysosomal route and not the proteasome pathway, because the narrow proteasome barrel precludes entry of oligomers and aggregates.
The FAP TTR Y114C mutant impaired autophagy in an in vitro cell model, whereas recovery of autophagy with curcumin decreased intracellular amounts of monomeric TTR [288]. Similarly, TTR V30M aggregates caused autophagy impairment in vivo in the gastrointestinal tract of transgenic mice, which could be reversed with autophagy inducers, which also reduced apoptosis [289]. These data suggest that impairment of autophagy is linked to the pathogenesis of TTR FAP. Although experimental proof for a role in the peripheral neuropathy in FAP is lacking, autophagy dysfunction is emerging as a common pathogenic mechanism in neuropathic pain [290,291]. Notably, a variety of genes mutated in inherited peripheral neuropathies are directly or indirectly associated with the autophagy pathway [292].
Efficient autophagy is key to clearing hIAPP oligomers, because in hIAPP transgenic mice hIAPP oligomers were more abundant in autophagy-deficient β-cells [293,294]. Similarly, pharmacological inhibition of autophagy in vitro promotes accumulation of IAPP aggregates and death of monkey islet cells expressing amyloidogenic simian IAPP (sIAPP), but not of rodent islet cells expressing non-amyloidogenic IAPP [293]. While autophagy insufficiency leads to accumulation of hIAPP oligomers and islet cell death, conversely hIAPP oligomers impair autophagy, as evidenced by accumulation of p62, a well-known autophagy substrate [295]. This feedback mechanism may lead to a profound decrease in autophagy and a large accumulation of hIAPP oligomers or islet amyloid. Thus, autophagy–lysosomal degradation defends β cells against proteotoxicity induced by oligomerization-prone human IAPP, and as such hIAPP-induced autophagy impairment may facilitate β-cell dysfunction in the development of T2DM (also see [296]). Oligomers and amyloid also affect autophagy in neurons. Aβ accumulation in neurons causes autophagy and mitophagy abnormalities, leading to neuronal dysfunction [297,298]. In the AD brain, defects in the autophagic–lysosomal pathway and accumulation of autophagic vacuoles in dystrophic neurites are observed [299], indicating Aβ-induced dysfunctional autophagy [300,301].
Dysregulation of autophagy in amyloid clearing cells affects not only the phagocytosis and degradation of amyloid and protein aggregates, but also the degradation of other autophagy substrates, such as damaged organelles, dead cells, synaptic material, and myelin debris. This reduced autophagy flux promotes an inflammatory phenotype of macrophages, microglia, and apoptosis [302]. Similarly, in amyloid-protein-producing cells, including neurons [255,256], impaired phagolysosomal degradation causes proinflammatory cytokine gene expression. These inflammatory mediators, notably IL-1β and IL-6, cause cell dysfunction, pyroptosis (inflammatory caspase-mediated cell death [303]), and tissue damage [304,305].
In conclusion, autophagy impairment by amyloid or oligomers is well documented in both neurodegenerative and metabolic amyloid diseases, and is even considered as a treatment option for these diseases [306]. For peripheral amyloid neuropathies, evidence of a role of autophagy impairment is still limited (only shown for TTR mutants) and should be further investigated.
4.7. Schwann Cells
A phenomenon that is often observed in familial amyloid neuropathy is defective myelination [91,307,308]. As such, impairment in Schwann cell function is emerging as an important mechanism in the development of amyloid neuropathy or neuropathic pain in general [309]. Schwann cells support the structural and functional integrity of peripheral nerves and are affected by the cellular toxicity of amyloid protein aggregates [75,90,310]. Schwann cells in contact with TTR amyloid fibrils become atrophic and distorted due to damage to their membranes [96,307]. Schwann cells also produce TTR, and in human TTRv transgenic mice TTR aggregates are present in the cytoplasm of Schwann cells. In addition, conditioned medium of these TTRv Schwann cells inhibited sensory neuron neurite outgrowth [311]. Thus, TTR gene expression in Schwann cells might contribute to neurodegeneration in FAP [91]. Schwann cells also produce SAA, the expression of which is upregulated by IL-6 and corticosteroids [312]. Therefore, SAA aggregates may damage Schwann cells when SAA is overproduced secondary to inflammatory diseases.
4.8. Microangiopathy
Amyloid protein aggregation impairs cell function through (dys)regulation of various intracellular processes, as already discussed. However, amyloid deposits may also affect cell function and cell survival by impairment of blood flow (microangiopathy), causing reduced supply of nutrients and oxygen. In hTTRv FAP patients, amyloid fibrils are present in extracellular spaces of the endoneurium, whereas amorphous deposits are present around micro vessels and in the subperineural space [313]. It has also been described that TTRv amyloid deposits cluster around endoneurial blood vessels and even invade their walls, with subsequent occlusion of endoneurial vessels themselves [307]. TTR aggregates also induce apoptosis of endothelial cells [75]. The TTR-induced endothelial cell damage and direct compression of blood vessels by amyloid deposits likely contribute to microangiopathy in FAP [313,314]. In hereditary gelsolin amyloidosis [76] and in AL amyloidosis [113,114,115], amyloid deposits are present in and around vascular walls of nerves. Similarly, in carpal tunnel amyloidosis in dialysis patients, β2M amyloid fibrils cause endothelial basement membrane enlargement and disruption and proliferation of endothelial cells [315].
In common amyloid diseases, amyloid also causes microangiopathy, which may contribute to neuropathy. SAA affects both aortic endothelial cell and vascular smooth muscle cell function [316,317] through activation of RAGE [316] o TLR2 [318]. Pharmacological inhibition of RAGE and TLR2/4 reduces human endothelial cell dysfunction [319]. In a mouse model of SAA amyloidosis, amyloid fibril accumulation associates with lesions in basal membranes; membranes of the endoplasmic reticulum; mitochondria; and the nucleus in endothelial cells in the kidney, liver, and spleen [315].
In both T2DM and AD, hIAPP amyloid or aggregates are found in and around blood vessels; however, until now no reports exist on hIAPP aggregates in the vasculature of the peripheral nervous system. Nevertheless, microangiopathy is a characteristic early feature of pancreatic islet pathology in human T2DM [320], which was also the case in a HIP rat model of T2DM, where hIAPP was overexpressed [320,321]. In agreement with this notion, amyloid deposition in the pancreatic islets of humans, monkeys, and cats begins in close proximity to islet capillaries [322].
In T2DM patients with dementia, hIAPP depositions are present in the brain microvasculature and cause endothelial dysfunction and vessel wall disruption. Similarly, diabetic HIP rats and rats injected intravenously with aggregated hIAPP develop endothelial dysfunction and vessel wall disruption associated with neurological deficits [323]. Such endothelial disruption may lead to increased vascular permeability. Indeed, patients with diabetic polyneuropathy show increased permeability of the blood–nerve barrier [324]. This increased permeability may promote the development of neuropathy due to various mechanisms, including endoneurial edema and endoneurial osmotic imbalance [325,326]. Overall, these data indicate that hIAPP-mediated angiopathy is present in the CNS and in the pancreatic islets in T2DM. Since hIAPP-related microangiopathy is also observed outside the location of hIAPP production and because hIAPP aggregates are not restricted to this location, hIAPP might also cause angiopathy within the PNS, facilitating the development of neuropathy.
Amyloid proteins might also impair blood flow indirectly by causing damage to the autonomic nervous system, impairing vascular regulation. In T2DM, autonomic dysfunction is observed and the Aβ protein impairs sympathetic innervation of multiple organs [327].
5. Summary and Conclusions
Aggregation of amyloid-fibril-forming proteins causes tissue dysregulation and damage by affecting different cell types, including amyloid-protein-producing cells, amyloid-clearing cells, vascular cells, and Schwann cells. The neurotoxic actions of amyloid and the dysregulation of these other cell types may contribute to an environment that promotes damage to neurons. Apparently, similar molecular and cell biological defects are caused by aggregation of the various amyloid proteins in known (usually rare) amyloid neuropathies and in some common diseases with amyloid and peripheral neuropathy, which are not (yet) considered as amyloid neuropathies. Intriguingly similar molecular and cell biological defects are also observed in models of neuropathy. Therefore, we propose that amyloid proteins are a common cause of peripheral neuropathy in these, and potentially other, diseases. Based on the current knowledge of pathogenic mechanisms involved in amyloid-protein-induced cell and tissue damage in various amyloidoses, we propose a “model” where these mechanisms are also at the root of “peripheral amyloid neuropathies”. Since experimental proof for the applicability of this model in common amyloid diseases with peripheral neuropathy is still scarce, more research into this field is required; even more so if one realizes that neuropathic pain affects 7–10% of the global population and that underlying mechanisms in the various types of painful peripheral neuropathy are still far from understood. Therefore, understanding if and how amyloid proteins contribute to peripheral neuropathy, particularly in common acquired diseases, is of high clinical and societal relevance and may open up new strategies to prevent or treat neuropathy in these diseases
Author Contributions
Conceptualization, N.E. and J.W.M.H.; Writing—original draft preparation: M.M.H.A., S.E.; Figures drawing and editing: S.E. and N.E.; writing—critical review and editing: N.E and J.W.M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded from King Abdulaziz City for Science and Technology (KACST), Saudi Arabia. Grant number 8091/10, to Mohammed M. H. Asiri.
Acknowledgments
The authors would like to thank Dr. W. van Hecke (UMC Utrecht, Department of Pathology) for providing the tissue section for Figure 3.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Collins, M.P.; Dyck, P.J.B. Peripheral Nervous System Involvement. Rare Diseases of the Immune System. In Anti-Neutrophil Cytoplasmic Antibody (ANCA) Associated Vasculitis; Sinico, R., Guillevin, L., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Primers 2019, 5, 41. [Google Scholar] [CrossRef]
- Javed, S.; Hayat, T.; Menon, L.; Alam, U.; Malik, R.A. Diabetic peripheral neuropathy in people with type 2 diabetes: Too little too late. Diabetic Med. 2019. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Barrell, K.; Smith, A.G. Peripheral Neuropathy. Med. Clin. N. Am. 2019, 103, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Katona, I.; Weis, J. Diseases of the peripheral nerves. Handb. Clin. Neurol. 2017, 145, 453–474. [Google Scholar] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018, 25, 215–219. [Google Scholar] [CrossRef]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef]
- Wechalekar, A.D.; Gillmore, J.D.; Hawkins, P.N. Systemic amyloidosis. Lancet 2016, 387, 2641–2654. [Google Scholar] [CrossRef]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2016 update on diagnosis, prognosis, and treatment. Am. J. Hematol. 2016, 91, 947–956. [Google Scholar] [CrossRef]
- Rambaran, R.N.; Serpell, L.C. Amyloid fibrils: Abnormal protein assembly. Prion 2008, 2, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.; Westermark, P. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014, 21, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Kisilevsky, R. The relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status, 2000. Amyloid 2000, 7, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Stefani, M.; Rigacci, S. Protein folding and aggregation into amyloid: The interference by natural phenolic compounds. Int. J. Mol. Sci. 2013, 14, 12411–12457. [Google Scholar] [CrossRef]
- Ferreira, S.T.; Vieira, M.N.; De Felice, F.G. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life 2007, 59, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 1952, 75, 408–427. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V. Transthyretin familial amyloid polyneuropathy: An update. J. Neurol. 2018, 265, 976–983. [Google Scholar] [CrossRef]
- Vital, C.; Vital, A.; Bouillot-Eimer, S.; Brechenmacher, C.; Ferrer, X.; Lagueny, A. Amyloid neuropathy: A retrospective study of 35 peripheral nerve biopsies. J. Peripher. Nerv. Syst. 2004, 9, 232–241. [Google Scholar] [CrossRef]
- Caldwell, J.H.; Klevanski, M.; Saar, M.; Muller, U.C. Roles of the amyloid precursor protein family in the peripheral nervous system. Mech. Dev. 2013, 130, 433–446. [Google Scholar] [CrossRef]
- Kanta, M.; Ehler, E.; Kohout, A.; Habalova, J.; Hanacek, R.; Vysata, O.; Brtkova, J.; Rehak, S.; Valis, M. Rare case of a localized radial nerve amyloid neuropathy. J. Clin. Neuromuscul. Dis. 2014, 16, 20–23. [Google Scholar] [CrossRef]
- Montealegre Sanchez, G.A.; Hashkes, P.J. Neurological manifestations of the Mendelian-inherited autoinflammatory syndromes. Dev. Med. Child Neurol. 2009, 51, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Real de Asua, D.; Costa, R.; Galvan, J.M.; Filigheddu, M.T.; Trujillo, D.; Cadinanos, J. Systemic AA amyloidosis: Epidemiology, diagnosis, and management. Clin. Epidemiol. 2014, 6, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Hoppener, J.W.; Ahren, B.; Lips, C.J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Hicks, C.W.; Selvin, E. Epidemiology of Peripheral Neuropathy and Lower Extremity Disease in Diabetes. Curr. Diabetes Rep. 2019, 19, 86. [Google Scholar] [CrossRef]
- Jolivalt, C.G.; Calcutt, N.A.; Masliah, E. Similar pattern of peripheral neuropathy in mouse models of type 1 diabetes and Alzheimer’s disease. Neuroscience 2012, 202, 405–412. [Google Scholar] [CrossRef]
- Grambalova, Z.; Calcutt, N.A.; Masliah, E. Peripheral neuropathy in Parkinson’s disease. Neuroendocrinol. Lett. 2015, 36, 363–367. [Google Scholar]
- Shin, S.C.; Robinson-Papp, J. Amyloid neuropathies. Mt. Sinai J. Med. 2012, 79, 733–748. [Google Scholar] [CrossRef]
- Gonzalez-Lopez, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; de Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef]
- Zhao, L.; Buxbaum, J.N.; Reixach, N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry 2013, 52, 1913–1926. [Google Scholar] [CrossRef]
- Eichner, T.; Radford, S.E. Understanding the complex mechanisms of beta2-microglobulin amyloid assembly. FEBS J. 2011, 278, 3868–3883. [Google Scholar] [CrossRef]
- Guo, J.T.; Yu, J.; Grass, D.; de Beer, F.C.; Kindy, M.S. Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J. Neurosci. 2002, 22, 5900–5909. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.P.; Al-Shawi, R.; Ellmerich, S.; Speck, I.; Aslam, S.; Hutchinson, W.L.; Mangione, P.P.; Disterer, P.; Gilbertson, J.A.; Hunt, T.; et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16115–16120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Huang, X.; Li, J. Light chain amyloidosis: Where are the light chains from and how they play their pathogenic role? Blood Rev. 2017, 31, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Mulder, H.; Ahren, B.; Sundler, F. Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am. J. Physiol. 1996, 271, E1008–E1014. [Google Scholar] [CrossRef] [PubMed]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 2011, 91, 795–826. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Lachmann, H.J.; Rowczenio, D.; Gilbertson, J.A.; Zeng, C.H.; Liu, Z.H.; Li, L.S.; Wechalekar, A.; Hawkins, P.N. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J. Am. Soc. Nephrol. 2009, 20, 444–451. [Google Scholar] [CrossRef]
- Cakar, A.; Durmus-Tekce, H.; Parman, Y. Familial Amyloid Polyneuropathy. Arch. Neuropsychiatry 2019, 56, 150–156. [Google Scholar] [CrossRef]
- Plante-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Waddington-Cruz, M.; Ackermann, E.J.; Polydefkis, M.; Heitner, S.B.; Dyck, P.J.; Barroso, F.A.; Wang, A.K.; Berk, J.L.; Dyck, P.J.B.; Monia, B.P.; et al. Hereditary transthyretin amyloidosis: Baseline characteristics of patients in the NEURO-TTR trial. Amyloid 2018, 25, 180–188. [Google Scholar] [CrossRef]
- Schmidt, H.H.; Waddington-Cruz, M.; Botteman, M.F.; Carter, J.A.; Chopra, A.S.; Hopps, M.; Stewart, M.; Fallet, S.; Amass, L. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve 2018, 57, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Kaku, M.; Berk, J.L. Neuropathy Associated with Systemic Amyloidosis. Semin. Neurol. 2019, 39, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Schonland, S.; Yumlu, S.; Hegenbart, U.; von Hutten, H.; Gioeva, Z.; Lohse, P.; Buttner, J.; Schmidt, H.; Rocken, C. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens: Identification of three novel mutations in the APOA1 gene. J. Mol. Diagn. 2009, 11, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 1998, 5, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, V.; Rasmussen, N.; Juhl, B.R.; Gimsing, P.; Vorstrup, S. Combined bilateral submandibular and sublingual swelling, macroglossus, and carpal tunnel syndrome caused by light chain amyloidosis. Ear Nose Throat J. 1998, 77, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2018 Update on diagnosis, prognosis, and treatment. Am. J. Hematol. 2018, 93, 1169–1180. [Google Scholar] [CrossRef]
- Quock, T.P.; Yan, T.; Chang, E.; Guthrie, S.; Broder, M.S. Epidemiology of AL amyloidosis: A real-world study using US claims data. Blood Adv. 2018, 2, 1046–1053. [Google Scholar] [CrossRef]
- Cheng, R.R.; Eskandari, R.; Welsh, C.T.; Varma, A.K. A case of isolated amyloid light-chain amyloidosis of the radial nerve. J. Neurosurg. 2016, 125, 598–602. [Google Scholar] [CrossRef]
- Floege, J.; Ketteler, M. beta2-microglobulin-derived amyloidosis: An update. Kidney Int. Suppl. 2001, 78, S164–S171. [Google Scholar] [CrossRef]
- Scarpioni, R.; Ricardi, M.; Albertazzi, V.; De Amicis, S.; Rastelli, F.; Zerbini, L. Dialysis-related amyloidosis: Challenges and solutions. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 319–328. [Google Scholar] [CrossRef]
- Pinney, J.H.; Whelan, C.J.; Petrie, A.; Dungu, J.; Banypersad, S.M.; Sattianayagam, P.; Wechalekar, A.; Gibbs, S.D.; Venner, C.P.; Wassef, N.; et al. Senile systemic amyloidosis: Clinical features at presentation and outcome. J. Am. Heart Assoc. 2013, 2, e000098. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Peuralinna, T.; Polvikoski, T.; Notkola, I.L.; Sulkava, R.; Hardy, J.; Singleton, A.; Kiuru-Enari, S.; Paetau, A.; Tienari, P.J.; et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 2008, 40, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Roman-Pintos, L.M.; Villegas-Rivera, G.; Rodriguez-Carrizalez, A.D.; Miranda-Diaz, A.G.; Cardona-Munoz, E.G. Diabetic Polyneuropathy in Type 2 Diabetes Mellitus: Inflammation, Oxidative Stress, and Mitochondrial Function. J. Diabetes Res. 2016, 2016, 3425617. [Google Scholar] [CrossRef] [PubMed]
- IDF Diabetes atlas 9th edition 2019. Available online: https://www.diabetesatlas.org/en/ (accessed on 26 June 2020).
- Sun, J.; Wang, Y.; Zhang, X.; Zhu, S.; He, H. Prevalence of peripheral neuropathy in patients with diabetes: A systematic review and meta-analysis. Prim. Care Diabetes 2020. [Google Scholar] [CrossRef]
- Juster-Switlyk, K.; Smith, A.G. Updates in diabetic peripheral neuropathy. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Kaeley, N.; Ahmad, S.; Pathania, M.; Kakkar, R. Prevalence and patterns of peripheral neuropathy in patients of rheumatoid arthritis. J. Fam. Med. Prim Care 2019, 8, 22–26. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef]
- Figueroa, J.J.; Loftus, E.V., Jr.; Harmsen, W.S.; Dyck, P.J.; Klein, C.J. Peripheral neuropathy incidence in inflammatory bowel disease: A population-based study. Neurology 2013, 80, 1693–1697. [Google Scholar] [CrossRef]
- Kim, S.; Kang, S.J.; Oh, K.W.; Ahn, B.K.; Lee, H.L.; Han, D.S.; Jang, K.; Kim, Y.S. Chronic inflammatory demyelinating polyneuropathy-like neuropathy as an initial presentation of Crohn’s disease. BMC Neurol. 2015, 15, 48. [Google Scholar] [CrossRef]
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr.; Shivashankar, R.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeiseter, A.R. Updated Incidence and Prevalence of Crohn’s Disease and Ulcerative Colitis in Olmsted County, Minnesota (1970–2011). ACG 2014 Ann. Sci. Meet. 2014, 15, 10. [Google Scholar]
- Garcia-Cabo, C.; Moris, G. Peripheral neuropathy: An underreported neurologic manifestation of inflammatory bowel disease. Eur. J. Intern. Med. 2015, 26, 468–475. [Google Scholar] [CrossRef]
- Moreton, B.J.; Tew, V.; das Nair, R.; Wheeler, M.; Walsh, D.A.; Lincoln, N.B. Pain phenotype in patients with knee osteoarthritis: Classification and measurement properties of painDETECT and self-report Leeds assessment of neuropathic symptoms and signs scale in a cross-sectional study. Arthritis Care Res. Hoboken 2015, 67, 519–528. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Prevalence, Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Bijlsma, J.W. Analgesia and the patient with osteoarthritis. Am. J. Ther. 2002, 9, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswami, P.; Chapman, K.M.; Yang, M.L.; Rutkove, S.B. Psoriatic arthritis-associated polyneuropathy: A report of three cases. J. Clin. Neuromuscul. Dis. 2007, 9, 248–251. [Google Scholar] [CrossRef]
- Scotti, L.; Franchi, M.; Marchesoni, A.; Corrao, G. Prevalence and incidence of psoriatic arthritis: A systematic review and meta-analysis. Semin. Arthritis Rheum. 2018, 48, 28–34. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Touitou, I. Familial mediterranean Fever in the world. Arthritis Rheum. 2009, 61, 1447–1453. [Google Scholar] [CrossRef]
- Ozdogan, H.; Ugurlu, S. Familial Mediterranean Fever. Presse Med. 2019, 48, e61–e76. [Google Scholar] [CrossRef]
- Tran, T.A. Muckle-Wells syndrome: Clinical perspectives. Open Access Rheumatol. 2017, 9, 123–129. [Google Scholar] [CrossRef]
- Sommer, C.; Schroder, J.M. Amyloid neuropathy: Immunocytochemical localization of intra- and extracellular immunoglobulin light chains. Acta Neuropathol. 1989, 79, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, K.; Fujimura, H.; Ueno, S.; Yoshikawa, H.; Kaido, M.; Nishimura, T.; Yorifuji, S.; Yanagihara, T. Familial amyloid polyneuropathy associated with transthyretin Gly42 mutation: A quantitative light and electron microscopic study of the peripheral nervous system. Acta Neuropathol. 1995, 90, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.M.; Du Yan, S.; Fernandes, R.; Guimaraes, A.; Stern, D.; Saraiva, M.J. Familial amyloid polyneuropathy: Receptor for advanced glycation end products-dependent triggering of neuronal inflammatory and apoptotic pathways. J. Neurosci. 2001, 21, 7576–7586. [Google Scholar] [CrossRef] [PubMed]
- Kiuru-Enari, S.; Somer, H.; Seppalainen, A.M.; Notkola, I.L.; Haltia, M. Neuromuscular pathology in hereditary gelsolin amyloidosis. J. Neuropathol. Exp. Neurol. 2002, 61, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.J.; Bosch, E.P.; Dyck, P.J.; Singer, W.; Vrana, J.A.; Theis, J.D.; Dogan, A.; Klein, C.J. Amyloid-like IgM deposition neuropathy: A distinct clinico-pathologic and proteomic profiled disorder. J. Peripher. Nerv. Syst. 2012, 17, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, B.L.; Garg, C.; Vasishta, R.K.; Nada, R.; Goyal, M.K. Localised SAA amyloidosis with intra-axonal intra-myelin amyloid deposits. Pathology 2017, 49, 103–105. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Correia, S.C.; Carvalho, C.; Moreira, P.I. Mitochondria in Alzheimer’s Disease and Diabetes-Associated Neurodegeneration: License to Heal! Handb. Exp. Pharmacol. 2017, 240, 281–308. [Google Scholar]
- Moreira, P.I. Sweet Mitochondria: A Shortcut to Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1391–1401. [Google Scholar] [CrossRef]
- Pivovarov, A.S.; Calahorro, F.; Walker, R.J. Na(+)/K(+)-pump and neurotransmitter membrane receptors. Invert. Neurosci. 2018, 19, 1. [Google Scholar] [CrossRef]
- Hazenberg, B.P. Amyloidosis: A clinical overview. Rheum. Dis. Clin. N. Am. 2013, 39, 323–345. [Google Scholar] [CrossRef]
- Loavenbruck, A.J.; Singer, W.; Mauermann, M.L.; Sandroni, P.; PJ, B.D.; Gertz, M.; Klein, C.J.; Low, P.A. Transthyretin amyloid neuropathy has earlier neural involvement but better prognosis than primary amyloid counterpart: An answer to the paradox? Ann. Neurol. 2016, 80, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Said, G.; Ropert, A.; Faux, N. Length-dependent degeneration of fibers in Portuguese amyloid polyneuropathy: A clinicopathologic study. Neurology 1984, 34, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Gertz, M.A.; Kyle, R.A. Prognosis of patients with primary systemic amyloidosis who present with dominant neuropathy. Am. J. Med. 1998, 104, 232–237. [Google Scholar] [CrossRef]
- Adams, D.; Lozeron, P.; Theaudin, M.; Denier, C.; Fagniez, O.; Rerat, K.; Signate, A.; Corcia, P.; Lacroix, C. Varied patterns of inaugural light-chain (AL) amyloid polyneuropathy: A monocentric study of 24 patients. Amyloid 2011, 18, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.; Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 2017, 105, 300–320. [Google Scholar] [CrossRef]
- Pareyson, D.; Saveri, P.; Sagnelli, A.; Piscosquito, G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci. Lett. 2015, 596, 66–77. [Google Scholar] [CrossRef]
- Simmons, Z.; Specht, C.S. The neuromuscular manifestations of amyloidosis. J. Clin. Neuromuscul. Dis. 2010, 11, 145–157. [Google Scholar] [CrossRef]
- Sousa, M.M.; Saraiva, M.J. Neurodegeneration in familial amyloid polyneuropathy: From pathology to molecular signaling. Prog. Neurobiol. 2003, 71, 385–400. [Google Scholar] [CrossRef]
- Murakami, T.; Sunada, Y. Transthyretin Amyloid Neuropathy: The Schwann Cell Hypothesis. Adv. Exp. Med. Biol. 2019, 1190, 371–378. [Google Scholar]
- Sekijima, Y.; Kelly, J.W.; Ikeda, S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr. Pharm. Des. 2008, 14, 3219–3230. [Google Scholar] [CrossRef]
- Wang, A.K.; Fealey, R.D.; Gehrking, T.L.; Low, P.A. Patterns of neuropathy and autonomic failure in patients with amyloidosis. Mayo Clin. Proc. 2008, 83, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, A.; Andrade, C. Familial amyloid polyneuropathy: An electron microscope study of the peripheral nerve in five cases. I. Interstitial changes. Brain 1971, 94, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Testro, A.G.; Brennan, S.O.; Macdonell, R.A.; Hawkins, P.N.; Angus, P.W. Hereditary amyloidosis with progressive peripheral neuropathy associated with apolipoprotein AI Gly26Arg: Outcome of hepatorenal transplantation. Liver Transpl. 2007, 13, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Ikeda, S.; Takahashi, M.; Kawagashira, Y.; Iijima, M.; Misumi, Y.; Ando, Y.; Ikeda, S.I.; Katsuno, M.; Sobue, G. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology 2016, 87, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.; Dogan, A.; Theis, J.D.; Vrana, J.A.; Lachmann, H.J.; Wechalekar, A.D.; Gilbertson, J.A.; Hunt, T.; Gibbs, S.D. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am. J. Pathol. 2011, 179, 1978–1987. [Google Scholar] [CrossRef]
- Paunio, T.; Kangas, H.; Kiuru, S.; Palo, J.; Peltonen, L.; Syvanen, A.C. Tissue distribution and levels of gelsolin mRNA in normal individuals and patients with gelsolin-related amyloidosis. FEBS Lett. 1997, 406, 49–55. [Google Scholar] [CrossRef]
- Burtnick, L.D.; Koepf, E.K.; Grimes, J.; Jones, E.Y.; Stuart, D.I.; McLaughlin, P.J.; Robinson, R.C. The crystal structure of plasma gelsolin: Implications for actin severing, capping, and nucleation. Cell 1997, 90, 661–670. [Google Scholar] [CrossRef]
- Bucki, R.; Levental, I.; Kulakowska, A.; Janmey, P.A. Plasma gelsolin: Function, prognostic value, and potential therapeutic use. Curr. Protein Pept. Sci. 2008, 9, 541–551. [Google Scholar] [CrossRef]
- Westberg, J.A.; Zhang, K.Z.; Andersson, L.C. Regulation of neural differentiation by normal and mutant (G654A, amyloidogenic) gelsolin. FASEB J. 1999, 13, 1621–1626. [Google Scholar] [CrossRef]
- Kyle, R.A.; Linos, A.; Beard, C.M.; Linke, R.P.; Gertz, M.A.; O’Fallon, W.M.; Kurland, L.T. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood 1992, 79, 1817–1822. [Google Scholar] [CrossRef]
- Schonland, S.O.; Hegenbart, U.; Bochtler, T.; Mangatter, A.; Hansberg, M.; Ho, A.D.; Lohse, P.; Rocken, C. Immunohistochemistry in the classification of systemic forms of amyloidosis: A systematic investigation of 117 patients. Blood 2012, 119, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Palladini, G. Light chain amyloidosis: The heart of the problem. Haematologica 2013, 98, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wang, Z.; Liu, H.; Liu, D.; Gong, Z.; Qi, J.; Hu, J. Clinical characteristics and diagnosis of a rare case of systemic AL amyloidosis: A descriptive study. Oncotarget 2018, 9, 24283–24290. [Google Scholar] [CrossRef] [PubMed]
- Heegaard, N.H. beta(2)-microglobulin: From physiology to amyloidosis. Amyloid 2009, 16, 151–173. [Google Scholar] [CrossRef]
- Haan, J.; Peters, W.G. Amyloid and peripheral nervous system disease. Clin. Neurol. Neurosurg. 1994, 96, 1–9. [Google Scholar] [CrossRef]
- O’Regan, J.; Walsh, R.; Kelly, D.; Plant, L.; Eustace, J. Neuropathy in the hemodialysis population: A review of neurophysiology referrals in a tertiary center. Ren. Fail. 2012, 34, 538–541. [Google Scholar] [CrossRef]
- Morris, A.D.; Smith, R.N.; Stone, J.R. The pathology and changing epidemiology of dialysis-related cardiac beta-2 microglobulin amyloidosis. Cardiovasc. Pathol. 2019, 42, 30–35. [Google Scholar] [CrossRef]
- Bodard, Q.; Roca, F.; Dilly, B.; Laurent, D.; Chassagne, P. Acute cardiac failure secondary to senile systemic amyloidosis. Age Ageing 2016, 45, 908–909. [Google Scholar] [CrossRef]
- Das, M.; Wilson, C.J.; Mei, X.; Wales, T.E.; Engen, J.R.; Gursky, O. Structural Stability and Local Dynamics in Disease-Causing Mutants of Human Apolipoprotein A-I: What Makes the Protein Amyloidogenic? J. Mol. Biol. 2016, 428, 449–462. [Google Scholar] [CrossRef]
- Hammarstrom, P.; Jiang, X.; Hurshman, A.R.; Powers, E.T.; Kelly, J.W. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc. Natl. Acad. Sci. USA 2002, 99, 16427–16432. [Google Scholar] [CrossRef]
- Russo, P.; Palladini, G.; Foli, A.; Zenone Bragotti, L.; Milani, P.; Nuvolone, M.; Obici, L.; Perfetti, V.; Brugnatelli, S.; Invernizzi, R.; et al. Liver involvement as the hallmark of aggressive disease in light chain amyloidosis: Distinctive clinical features and role of light chain type in 225 patients. Amyloid 2011, 18, 92–93. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Katoh, N.; Ikeda, S. Clinical manifestations at diagnosis in Japanese patients with systemic AL amyloidosis: A retrospective study of 202 cases with a special attention to uncommon symptoms. Intern. Med. 2014, 53, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Palladini, G.; Hegenbart, U.; Milani, P.; Kimmich, C.; Foli, A.; Ho, A.D.; Vidus Rosin, M.; Albertini, R.; Moratti, R.; Merlini, G.; et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014, 124, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Gillmore, J.D.; Wechalekar, A.; Bird, J.; Cavenagh, J.; Hawkins, S.; Kazmi, M.; Lachmann, H.J.; Hawkins, P.N.; Pratt, G.; the BCSH Committee. Guidelines on the diagnosis and investigation of AL amyloidosis. Br. J. Haematol. 2015, 168, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkanen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983, 75, 618–623. [Google Scholar] [CrossRef]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. Lausanne 2017, 8, 343. [Google Scholar] [CrossRef]
- Cernea, S.; Dobreanu, M. Diabetes and beta cell function: From mechanisms to evaluation and clinical implications. Biochem. Med. Zagreb 2013, 23, 266–280. [Google Scholar] [CrossRef]
- Halban, P.A.; Polonsky, K.S.; Bowden, D.W.; Hawkins, M.A.; Ling, C.; Mather, K.J.; Powers, A.C.; Rhodes, C.J.; Sussel, L.; Weir, G.C. beta-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care 2014, 37, 1751–1758. [Google Scholar] [CrossRef]
- Saisho, Y. beta-cell dysfunction: Its critical role in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 109–124. [Google Scholar] [CrossRef]
- Beery, M.L.; Jacobsen, L.M.; Atkinson, M.A.; Butler, A.E.; Campbell-Thompson, M. Islet amyloidosis in a child with type 1 diabetes. Islets 2019, 11, 44–49. [Google Scholar] [CrossRef]
- Abbott, C.A.; Malik, R.A.; van Ross, E.R.; Kulkarni, J.; Boulton, A.J. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care 2011, 34, 2220–2224. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Sharma, N.; Pasnoor, M.; Kluding, P.M. Painful Diabetic Peripheral Neuropathy: Presentations, Mechanisms, and Exercise Therapy. J. Diabetes Metab. 2013. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.A. Diabetic neuropathy: A focus on small fibres. Diabetes Metab. Res. Rev. 2019, 36, e3255. [Google Scholar] [CrossRef] [PubMed]
- Shillo, P.; Sloan, G.; Greig, M.; Hunt, L.; Selvarajah, D.; Elliott, J.; Gandhi, R.; Wilkinson, I.D.; Tesfaye, S. Painful and Painless Diabetic Neuropathies: What Is the Difference? Curr. Diabetes Rep. 2019, 19, 32. [Google Scholar] [CrossRef]
- Selvarajah, D.; Kar, D.; Khunti, K.; Davies, M.J.; Scott, A.R.; Walker, J.; Tesfaye, S. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019, 7, 938–948. [Google Scholar] [CrossRef]
- Premkumar, L.S.; Pabbidi, R.M. Diabetic peripheral neuropathy: Role of reactive oxygen and nitrogen species. Cell Biochem. Biophys. 2013, 67, 373–383. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef]
- Shanmugam, N.; Reddy, M.A.; Guha, M.; Natarajan, R. High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 2003, 52, 1256–1264. [Google Scholar] [CrossRef]
- Figueroa-Romero, C.; Sadidi, M.; Feldman, E.L. Mechanisms of disease: The oxidative stress theory of diabetic neuropathy. Rev. Endocr. Metab. Disord. 2008, 9, 301–314. [Google Scholar] [CrossRef]
- Zochodne, D.W. Diabetic polyneuropathy: An update. Curr. Opin. Neurol. 2008, 21, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macri, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Lee, C.C.; Perkins, B.A.; Kayaniyil, S.; Harris, S.B.; Retnakaran, R.; Gerstein, H.C.; Zinman, B.; Hanley, A.J. Peripheral Neuropathy and Nerve Dysfunction in Individuals at High Risk for Type 2 Diabetes: The PROMISE Cohort. Diabetes Care 2015, 38, 793–800. [Google Scholar] [CrossRef]
- Grisold, A.; Callaghan, B.C.; Feldman, E.L. Mediators of diabetic neuropathy: Is hyperglycemia the only culprit? Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 103–111. [Google Scholar] [CrossRef]
- Hoppener, J.W.; Oosterwijk, C.; van Hulst, K.L.; Verbeek, J.S.; Capel, P.J.; de Koning, E.J.; Clark, A.; Jansz, H.S.; Lips, C.J. Molecular physiology of the islet amyloid polypeptide (IAPP)/amylin gene in man, rat, and transgenic mice. J. Cell Biochem. 1994, 55, 39–53. [Google Scholar] [CrossRef]
- Kiriyama, Y.; Nochi, H. Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet beta-Cells from Amylin Cytotoxicity. Cells 2018, 7, 95. [Google Scholar] [CrossRef]
- Dhanvantari, S. Unfolding the mechanisms of disease progression in permanent neonatal diabetes. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E383–E384. [Google Scholar] [CrossRef]
- Hoppener, J.W.; Oosterwijk, C.; Nieuwenhuis, M.G.; Posthuma, G.; Thijssen, J.H.; Vroom, T.M.; Ahren, B.; Lips, C.J. Extensive islet amyloid formation is induced by development of Type II diabetes mellitus and contributes to its progression: Pathogenesis of diabetes in a mouse model. Diabetologia 1999, 42, 427–434. [Google Scholar] [CrossRef]
- Chakraborty, S.; Mukherjee, B.; Basu, S. Pinpointing proline substitution to be responsible for the loss of amyloidogenesis in IAPP. Chem. Biol. Drug Des. 2013, 82, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engstrom, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Butler, P.C. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 2006, 47, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Liu, Z.H.; Zeng, C.H.; Peng, A.; Chen, H.P.; Zhou, H.; Li, L.S. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int. 2007, 72, 213–218. [Google Scholar] [CrossRef]
- Jackson, K.; Barisone, G.A.; Diaz, E.; Jin, L.W.; DeCarli, C.; Despa, F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann. Neurol. 2013, 74, 517–526. [Google Scholar] [CrossRef]
- Liu, M.; Verma, N.; Peng, X.; Srodulski, S.; Morris, A.; Chow, M.; Hersh, L.B.; Chen, J.; Zhu, H.; Netea, M.G.; et al. Hyperamylinemia Increases IL-1beta Synthesis in the Heart via Peroxidative Sarcolemmal Injury. Diabetes 2016, 65, 2772–2783. [Google Scholar] [CrossRef]
- Mulder, H.; Leckstrom, A.; Uddman, R.; Ekblad, E.; Westermark, P.; Sundler, F. Islet amyloid polypeptide (amylin) is expressed in sensory neurons. J. Neurosci. 1995, 15, 7625–7632. [Google Scholar] [CrossRef]
- Mulder, H.; Zhang, Y.; Danielsen, N.; Sundler, F. Islet amyloid polypeptide and calcitonin gene-related peptide expression are upregulated in lumbar dorsal root ganglia after unilateral adjuvant-induced inflammation in the rat paw. Brain Res. Mol. Brain Res. 1997, 50, 127–135. [Google Scholar] [CrossRef]
- Mulder, H.; Zhang, Y.; Danielsen, N.; Sundler, F. Islet amyloid polypeptide and calcitonin gene-related peptide expression are down-regulated in dorsal root ganglia upon sciatic nerve transection. Brain Res. Mol. Brain Res. 1997, 47, 322–330. [Google Scholar] [CrossRef]
- Gebre-Medhin, S.; Mulder, H.; Pekny, M.; Westermark, G.; Tornell, J.; Westermark, P.; Sundler, F.; Ahren, B. Increased insulin secretion and glucose tolerance in mice lacking islet amyloid polypeptide (amylin). Biochem. Biophys. Res. Commun. 1998, 250, 271–277. [Google Scholar] [CrossRef]
- Mulder, H.; Jongsma, H.; Zhang, Y.; Gebre-Medhin, S.; Sundler, F.; Danielsen, N. Pituitary adenylate cyclase-activating polypeptide and islet amyloid polypeptide in primary sensory neurons: Functional implications from plasticity in expression on nerve injury and inflammation. Mol. Neurobiol. 1999, 19, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.S.; Castro-Lopes, J.M.; Neto, F.L.; Potes, C.S. Amylin, a peptide expressed by nociceptors, modulates chronic neuropathic pain. Eur. J. Pain 2019, 23, 784–799. [Google Scholar] [CrossRef] [PubMed]
- Gebre-Medhin, S.; Mulder, H.; Zhang, Y.; Sundler, F.; Betsholtz, C. Reduced nociceptive behavior in islet amyloid polypeptide (amylin) knockout mice. Mol. Brain Res. 1998, 63, 180–183. [Google Scholar] [CrossRef]
- Asiri, M.M.H.; Versteeg, S.; Höppener, J.W.M.; Eijkelkamp, N. Human IAPP mediates painful diabetic peripheral neuropathy. In Proceedings of the The 11th Congress of the European Pain Federation EFIC, Valencia, Spain, 4–7 September 2019. [Google Scholar]
- Hoppener, J.W.; Verbeek, J.S.; de Koning, E.J.; Oosterwijk, C.; van Hulst, K.L.; Visser-Vernooy, H.J.; Hofhuis, F.M.; van Gaalen, S.; Berends, M.J.; Hackeng, W.H.; et al. Chronic overproduction of islet amyloid polypeptide/amylin in transgenic mice: Lysosomal localization of human islet amyloid polypeptide and lack of marked hyperglycaemia or hyperinsulinaemia. Diabetologia 1993, 36, 1258–1265. [Google Scholar] [CrossRef] [PubMed]
- Potes, C.S.; Pestana, A.C.; Pontes, M.; Caramelo, A.S.; Neto, F.L. Amylin modulates the formalin-induced tonic pain behaviours in rats. Eur. J. Pain 2016, 20, 1741–1752. [Google Scholar] [CrossRef]
- Verma, N.; Ly, H.; Liu, M.; Chen, J.; Zhu, H.; Chow, M.; Hersh, L.B.; Despa, F. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1beta Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J. Alzheimers Dis. 2016, 53, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Srodulski, S.; Sharma, S.; Bachstetter, A.B.; Brelsfoard, J.M.; Pascual, C.; Xie, X.S.; Saatman, K.E.; Van Eldik, L.J.; Despa, F. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol. Neurodegener. 2014, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Westermark, G.T.; Fandrich, M.; Westermark, P. AA amyloidosis: Pathogenesis and targeted therapy. Annu. Rev. Pathol. 2015, 10, 321–344. [Google Scholar] [CrossRef]
- Papa, R.; Lachmann, H.J. Secondary, AA, Amyloidosis. Rheum. Dis. Clin. N. Am. 2018, 44, 585–603. [Google Scholar] [CrossRef]
- Agarwal, V.; Lachmann, H.J. A clinical, electrophysiological, and pathological study of neuropathy in rheumatoid arthritis. Clin. Rheumatol. 2008, 27, 841–844. [Google Scholar] [CrossRef]
- Akasaki, Y.; Reixach, N.; Matsuzaki, T.; Alvarez-Garcia, O.; Olmer, M.; Iwamoto, Y.; Buxbaum, J.N.; Lotz, M.K. Transthyretin deposition in articular cartilage: A novel mechanism in the pathogenesis of osteoarthritis. Arthritis Rheumatol. 2015, 67, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, A.; Ueda, M.; Sueyoshi, T.; Nakamura, E.; Tasaki, M.; Suenaga, G.; Motokawa, H.; Toyoshima, R.; Kinoshita, Y.; Misumi, Y.; et al. Knee osteoarthritis associated with different kinds of amyloid deposits and the impact of aging on type of amyloid. Amyloid 2016, 23, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bergis, M.; Dega, H.; Planquois, V.; Benichou, O.; Dubertret, L. Amyloidosis complicating psoriatic arthritis. Ann. Dermatol. Venereol. 2003, 130, 1039–1042. [Google Scholar] [PubMed]
- Basturk, T.; Ozagari, A.; Ozturk, T.; Kusaslan, R.; Unsal, A. Crohn’s disease and secondary amyloidosis: Early complication? A case report and review of the literature. J. Ren. Care 2009, 35, 147–150. [Google Scholar] [CrossRef]
- Pukitis, A.; Zake, T.; Groma, V.; Ostrovskis, E.; Skuja, S.; Pokrotnieks, J. Effect of infliximab induction therapy on secondary systemic amyloidosis associated with Crohn’s disease: Case report and review of the literature. J. Gastrointestin Liver Dis. 2013, 22, 333–336. [Google Scholar]
- Tada, Y.; Ishihara, S.; Ito, T.; Matsui, K.; Sonoyama, H.; Oka, A.; Kusunoki, R.; Fukuba, N.; Mishima, Y.; Oshima, N.; et al. Successful use of maintenance infliximab for nephropathy in a patient with secondary amyloidosis complicating Crohn’s disease. Intern. Med. 2013, 52, 1899–1902. [Google Scholar] [CrossRef]
- Vahdat Shariat Panahi, A.; Hultman, P.; Ollinger, K.; Westermark, G.T.; Lundmark, K. Lipid membranes accelerate amyloid formation in the mouse model of AA amyloidosis. Amyloid 2019, 26, 34–44. [Google Scholar] [CrossRef]
- Tanaka, M.; Nishimura, A.; Takeshita, H.; Takase, H.; Yamada, T.; Mukai, T. Effect of lipid environment on amyloid fibril formation of human serum amyloid A. Chem. Phys. Lipids 2017, 202, 6–12. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Fandrich, M.; Gursky, O. Serum amyloid A sequesters diverse phospholipids and their hydrolytic products, hampering fibril formation and proteolysis in a lipid-dependent manner. Chem. Commun. Camb. 2018, 54, 3532–3535. [Google Scholar] [CrossRef]
- Morgado, I.; Garvey, M. Lipids in Amyloid-beta Processing, Aggregation, and Toxicity. Adv. Exp. Med. Biol. 2015, 855, 67–94. [Google Scholar]
- Qiang, W.; Yau, W.M.; Schulte, J. Fibrillation of beta amyloid peptides in the presence of phospholipid bilayers and the consequent membrane disruption. Biochim. Biophys. Acta 2015, 1848, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoppener, J.W. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ren, B.; Liu, Y.; Liang, G.; Sun, Y.; Xu, L.; Zheng, J. Membrane Interactions of hIAPP Monomer and Oligomer with Lipid Membranes by Molecular Dynamics Simulations. ACS Chem. Neurosci. 2017, 8, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.M.; Mok, Y.F.; Howlett, G.J.; Griffin, M.D. The Role of Lipid in Misfolding and Amyloid Fibril Formation by Apolipoprotein C-II. Adv. Exp. Med. Biol. 2015, 855, 157–174. [Google Scholar] [PubMed]
- Bram, Y.; Frydman-Marom, A.; Yanai, I.; Gilead, S.; Shaltiel-Karyo, R.; Amdursky, N.; Gazit, E. Apoptosis induced by islet amyloid polypeptide soluble oligomers is neutralized by diabetes-associated specific antibodies. Sci. Rep. 2014, 4, 4267. [Google Scholar] [CrossRef]
- Kumar, S.; Birol, M.; Miranker, A.D. Foldamer scaffolds suggest distinct structures are associated with alternative gains-of-function in a preamyloid toxin. Chem. Commun. Camb. 2016, 52, 6391–6394. [Google Scholar] [CrossRef]
- Jayaraman, S.; Gantz, D.L.; Haupt, C.; Gursky, O. Serum amyloid A forms stable oligomers that disrupt vesicles at lysosomal pH and contribute to the pathogenesis of reactive amyloidosis. Proc. Natl. Acad. Sci. USA 2017, 114, E6507–E6515. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Tempra, C.; Scollo, F.; Milardi, D.; La Rosa, C. Amyloid growth and membrane damage: Current themes and emerging perspectives from theory and experiments on Abeta and hIAPP. Biochim. Biophys. Acta Biomembr. 2018. [Google Scholar] [CrossRef]
- Rawat, A.; Langen, R.; Varkey, J. Membranes as modulators of amyloid protein misfolding and target of toxicity. Biochim. Biophys. Acta Biomembr. 2018. [Google Scholar] [CrossRef]
- Fabiani, C.; Antollini, S.S. Alzheimer’s Disease as a Membrane Disorder: Spatial Cross-Talk Among Beta-Amyloid Peptides, Nicotinic Acetylcholine Receptors and Lipid Rafts. Front. Cell Neurosci. 2019, 13, 309. [Google Scholar] [CrossRef]
- Kagan, B.L.; Thundimadathil, J. Amyloid peptide pores and the beta sheet conformation. Adv. Exp. Med. Biol. 2010, 677, 150–167. [Google Scholar] [PubMed]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—Protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar] [PubMed]
- Gurlo, T.; Ryazantsev, S.; Huang, C.J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for proteotoxicity in beta cells in type 2 diabetes: Toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 2010, 176, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and Processing of the Amyloid-beta Protein Precursor in Mitochondria-Associated Membranes. J. Alzheimers Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ueda, Y.; Nakajima, A.; Mitsuyama, Y. EPR evidence of hydroxyl radical generation as an initiator of lipid peroxidation in amyloid beta-protein-stimulated PC12 cells. Brain Res. 2004, 1025, 29–34. [Google Scholar] [CrossRef]
- Shoeb, M.; Ansari, N.H.; Srivastava, S.K.; Ramana, K.V. 4-Hydroxynonenal in the pathogenesis and progression of human diseases. Curr. Med. Chem. 2014, 21, 230–237. [Google Scholar] [CrossRef]
- Gunn, A.P.; Wong, B.X.; Johanssen, T.; Griffith, J.C.; Masters, C.L.; Bush, A.I.; Barnham, K.J.; Duce, J.A.; Cherny, R.A. Amyloid-beta Peptide Abeta3pE-42 Induces Lipid Peroxidation, Membrane Permeabilization, and Calcium Influx in Neurons. J. Biol. Chem. 2016, 291, 6134–6145. [Google Scholar] [CrossRef]
- Hassler, S.N.; Johnson, K.M.; Hulsebosch, C.E. Reactive oxygen species and lipid peroxidation inhibitors reduce mechanical sensitivity in a chronic neuropathic pain model of spinal cord injury in rats. J. Neurochem. 2014, 131, 413–417. [Google Scholar] [CrossRef]
- Chadwick, S.R.; Lajoie, P. Endoplasmic Reticulum Stress Coping Mechanisms and Lifespan Regulation in Health and Diseases. Front. Cell Dev. Biol. 2019, 7, 84. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Ogen-Shtern, N.; Ben David, T.; Lederkremer, G.Z. Protein aggregation and ER stress. Brain Res. 2016, 1648, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Genereux, J.C.; Wiseman, R.L. Endoplasmic reticulum quality control and systemic amyloid disease: Impacting protein stability from the inside out. IUBMB Life 2015, 67, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.F.; Cerca, F.; Santos, S.D.; Saraiva, M.J. Endoplasmic reticulum stress associated with extracellular aggregates. Evidence from transthyretin deposition in familial amyloid polyneuropathy. J. Biol. Chem. 2006, 281, 21998–22003. [Google Scholar] [CrossRef]
- Casas, S.; Gomis, R.; Gribble, F.M.; Altirriba, J.; Knuutila, S.; Novials, A. Impairment of the ubiquitin-proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet amyloid polypeptide and contributes to pancreatic beta-cell apoptosis. Diabetes 2007, 56, 2284–2294. [Google Scholar] [CrossRef]
- Huang, C.J.; Lin, C.Y.; Haataja, L.; Gurlo, T.; Butler, A.E.; Rizza, R.A.; Butler, P.C. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 2007, 56, 2016–2027. [Google Scholar] [CrossRef]
- Matveyenko, A.V.; Gurlo, T.; Daval, M.; Butler, A.E.; Butler, P.C. Successful versus failed adaptation to high-fat diet-induced insulin resistance: The role of IAPP-induced beta-cell endoplasmic reticulum stress. Diabetes 2009, 58, 906–916. [Google Scholar] [CrossRef]
- Cadavez, L.; Montane, J.; Alcarraz-Vizan, G.; Visa, M.; Vidal-Fabrega, L.; Servitja, J.M.; Novials, A. Chaperones ameliorate beta cell dysfunction associated with human islet amyloid polypeptide overexpression. PLoS ONE 2014, 9, e101797. [Google Scholar] [CrossRef]
- Law, C.J.; Ashcroft, H.A.; Zheng, W.; Sexton, J.Z. Assay development and multivariate scoring for high-content discovery of chemoprotectants of endoplasmic-reticulum-stress-mediated amylin-induced cytotoxicity in pancreatic beta cells. Assay Drug Dev. Technol. 2014, 12, 375–384. [Google Scholar] [CrossRef]
- Lee, S.J.; Kang, H.K.; Eum, W.S.; Park, J.; Choi, S.Y.; Kwon, H.Y. Tat-biliverdin reductase A protects INS-1 cells from human islet amyloid polypeptide-induced cytotoxicity by alleviating oxidative stress and ER stress. Cell Biol. Int. 2017, 41, 514–524. [Google Scholar] [CrossRef]
- He, Y.M.; Zhang, Q.; Zheng, M.; Fan, Z.H.; Li, Y.H.; Zhang, D.; Zhang, Z.; Yuan, S.S.; Wang, Y.Y.; Zhou, P.; et al. Protective effects of a G. lucidum proteoglycan on INS-1 cells against IAPP-induced apoptosis via attenuating endoplasmic reticulum stress and modulating CHOP/JNK pathways. Int. J. Biol. Macromol. 2018, 106, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, K.; Tomiyama, T.; Ishibashi, K.; Ito, K.; Teraoka, R.; Lambert, M.P.; Klein, W.L.; Mori, H. The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am. J. Pathol. 2009, 174, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.E.; Ferreira, S.T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 2018, 136, 350–360. [Google Scholar] [CrossRef]
- Lupachyk, S.; Watcho, P.; Obrosov, A.A.; Stavniichuk, R.; Obrosova, I.G. Endoplasmic reticulum stress contributes to prediabetic peripheral neuropathy. Exp. Neurol. 2013, 247, 342–348. [Google Scholar] [CrossRef]
- O’Brien, P.D.; Hinder, L.M.; Sakowski, S.A.; Feldman, E.L. ER stress in diabetic peripheral neuropathy: A new therapeutic target. Antioxid. Redox. Signal. 2014, 21, 621–633. [Google Scholar]
- Inceoglu, B.; Bettaieb, A.; Trindade da Silva, C.A.; Lee, K.S.; Haj, F.G.; Hammock, B.D. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef]
- Lupachyk, S.; Watcho, P.; Stavniichuk, R.; Shevalye, H.; Obrosova, I.G. Endoplasmic reticulum stress plays a key role in the pathogenesis of diabetic peripheral neuropathy. Diabetes 2013, 62, 944–952. [Google Scholar] [CrossRef]
- Valenzuela, V.; Onate, M.; Hetz, C.; Court, F.A. Injury to the nervous system: A look into the ER. Brain Res. 2016, 1648, 617–625. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Feno, S.; Butera, G.; Vecellio Reane, D.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid. Med. Cell Longev. 2019, 2019, 9324018. [Google Scholar] [CrossRef] [PubMed]
- Rumora, A.E.; Savelieff, M.G.; Sakowski, S.A.; Feldman, E.L. Disorders of mitochondrial dynamics in peripheral neuropathy: Clues from hereditary neuropathy and diabetes. Int. Rev. Neurobiol. 2019, 145, 127–176. [Google Scholar] [PubMed]
- Trecarichi, A.; Flatters, S.J.L. Mitochondrial dysfunction in the pathogenesis of chemotherapy-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 83–126. [Google Scholar]
- Olsson, M.; Hellman, U.; Plante-Bordeneuve, V.; Jonasson, J.; Lang, K.; Suhr, O.B. Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin. Genet. 2009, 75, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bonaiti, B.; Olsson, M.; Hellman, U.; Suhr, O.; Bonaiti-Pellie, C.; Plante-Bordeneuve, V. TTR familial amyloid polyneuropathy: Does a mitochondrial polymorphism entirely explain the parent-of-origin difference in penetrance? Eur. J. Hum. Genet. 2010, 18, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: An organ-based analysis. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E268–E285. [Google Scholar] [CrossRef]
- Akhter, F.; Chen, D.; Yan, S.F.; Yan, S.S. Mitochondrial Perturbation in Alzheimer’s Disease and Diabetes. Prog. Mol. Biol. Transl. Sci. 2017, 146, 341–361. [Google Scholar]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef]
- Macdonald, R.; Barnes, K.; Hastings, C.; Mortiboys, H. Mitochondrial abnormalities in Parkinson’s disease and Alzheimer’s disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 2018, 46, 891–909. [Google Scholar] [CrossRef]
- Lim, Y.A.; Rhein, V.; Baysang, G.; Meier, F.; Poljak, A.; Raftery, M.J.; Guilhaus, M.; Ittner, L.M.; Eckert, A.; Gotz, J. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics 2010, 10, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Birol, M.; Kumar, S.; Rhoades, E.; Miranker, A.D. Conformational switching within dynamic oligomers underpins toxic gain-of-function by diabetes-associated amyloid. Nat. Commun. 2018, 9, 1312. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Hernando-Perez, E.; Nunez, L.; Villalobos, C. Amyloid beta Oligomers Increase ER-Mitochondria Ca(2+) Cross Talk in Young Hippocampal Neurons and Exacerbate Aging-Induced Intracellular Ca(2+) Remodeling. Front. Cell Neurosci. 2019, 13, 22. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schutz, O.; et al. alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Guimaraes-Costa, A.B.; Bandeira-Melo, C.; Chimelli, L.; Waddington-Cruz, M.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. Inflammatory profiling of patients with familial amyloid polyneuropathy. BMC Neurol. 2019, 19, 146. [Google Scholar] [CrossRef]
- Abedini, A.; Derk, J.; Schmidt, A.M. The receptor for advanced glycation endproducts is a mediator of toxicity by IAPP and other proteotoxic aggregates: Establishing and exploiting common ground for novel amyloidosis therapies. Protein Sci. 2018, 27, 1166–1180. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Othman, I.; Aamir, K.; Shaikh, M.F. Impact of HMGB1, RAGE, and TLR4 in Alzheimer’s Disease (AD): From Risk Factors to Therapeutic Targeting. Cells 2020, 9, 383. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflamm. 2013, 2013, 403460. [Google Scholar] [CrossRef]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- Sousa, M.M.; Yan, S.D.; Stern, D.; Saraiva, M.J. Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Investig. 2000, 80, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.L.; Yan, S.F.; Wendt, T.; Hans, D.; Pachydaki, S.; Bucciarelli, L.G.; Adebayo, A.; Qu, W.; Lu, Y.; Kostov, K.; et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004, 18, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.; Smith, D.R.; Tessler, L.; Mateo, A.R.; Martens, C.; Schartner, E.; Van der Ploeg, R.; Toth, C.; Zochodne, D.W.; Fernyhough, P. Receptor for advanced glycation end-products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp. Neurol. 2013, 249, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Brederson, J.D.; Strakhova, M.; Mills, C.; Barlow, E.; Meyer, A.; Nimmrich, V.; Leddy, M.; Simler, G.; Schmidt, M.; Jarvis, M.; et al. A monoclonal antibody against the receptor for advanced glycation end products attenuates inflammatory and neuropathic pain in the mouse. Eur. J. Pain 2016, 20, 607–614. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef]
- Sommer, C.; Leinders, M.; Uceyler, N. Inflammation in the pathophysiology of neuropathic pain. Pain 2018, 159, 595–602. [Google Scholar] [CrossRef]
- Safieh-Garabedian, B.; Nomikos, M.; Saade, N. Targeting inflammatory components in neuropathic pain: The analgesic effect of thymulin related peptide. Neurosci. Lett. 2019, 702, 61–65. [Google Scholar] [CrossRef]
- Raoof, R.; Willemen, H.; Eijkelkamp, N. Divergent roles of immune cells and their mediators in pain. Rheumatology 2018, 57, 429–440. [Google Scholar] [CrossRef]
- Coll, R.C.; O’Neill, L.; Schroder, K. Questions and controversies in innate immune research: What is the physiological role of NLRP3? Cell Death Discov. 2016, 2, 16019. [Google Scholar] [CrossRef]
- Migita, K.; Izumi, Y.; Jiuchi, Y.; Kozuru, H.; Kawahara, C.; Nakamura, M.; Nakamura, T.; Agematsu, K.; Masumoto, J.; Yasunami, M.; et al. Serum amyloid A induces NLRP-3-mediated IL-1beta secretion in neutrophils. PLoS ONE 2014, 9, e96703. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Liu, S.; Yi, X.; Zhang, S.; Ding, Y. Serum amyloid A induces interleukin-1beta secretion from keratinocytes via the NACHT, LRR and PYD domains-containing protein 3 inflammasome. Clin. Exp. Immunol. 2015, 179, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Song, L.T.; Lai, W.; Li, J.S.; Mu, Y.Z.; Li, C.Y.; Jiang, S.Y. The interaction between serum amyloid A and Toll-like receptor 2 pathway regulates inflammatory cytokine secretion in human gingival fibroblasts. J. Periodontol. 2020, 91, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef]
- Abedini, A.; Cao, P.; Plesner, A.; Zhang, J.; He, M.; Derk, J.; Patil, S.A.; Rosario, R.; Lonier, J.; Song, F.; et al. RAGE binds preamyloid IAPP intermediates and mediates pancreatic beta cell proteotoxicity. J. Clin. Investig. 2018, 128, 682–698. [Google Scholar] [CrossRef]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes 2014, 63, 1698–1711. [Google Scholar] [CrossRef]
- Park, Y.J.; Warnock, G.L.; Ao, Z.; Safikhan, N.; Meloche, M.; Asadi, A.; Kieffer, T.J.; Marzban, L. Dual role of interleukin-1beta in islet amyloid formation and its beta-cell toxicity: Implications for type 2 diabetes and islet transplantation. Diabetes Obes. Metab. 2017, 19, 682–694. [Google Scholar] [CrossRef]
- Mu, Z.P.; Wang, Y.G.; Li, C.Q.; Lv, W.S.; Wang, B.; Jing, Z.H.; Song, X.J.; Lun, Y.; Qiu, M.Y.; Ma, X.L. Association Between Tumor Necrosis Factor-alpha and Diabetic Peripheral Neuropathy in Patients with Type 2 Diabetes: A Meta-Analysis. Mol. Neurobiol. 2017, 54, 983–996. [Google Scholar] [CrossRef]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The Role of Neuronal NLRP1 Inflammasome in Alzheimer’s Disease: Bringing Neurons into the Neuroinflammation Game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef] [PubMed]
- Nan, K.; Han, Y.; Fang, Q.; Huang, C.; Yu, L.; Ge, W.; Xiang, F.; Tao, Y.X.; Cao, H.; Li, J. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-kappaB signaling in primary hippocampal neurons induced by Abeta25-35. Int. Immunopharmacol. 2019, 67, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lathia, J.D.; Selvaraj, P.K.; Jo, D.G.; Mughal, M.R.; Cheng, A.; Siler, D.A.; Markesbery, W.R.; Arumugam, T.V.; Mattson, M.P. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 2008, 213, 114–121. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Moyse, E.; Haddad, M.; Benlabiod, C.; Ramassamy, C.; Krantic, S. Common Pathological Mechanisms and Risk Factors for Alzheimer’s Disease and Type-2 Diabetes: Focus on Inflammation. Curr. Alzheimer Res. 2019, 16, 986–1006. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimers Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef]
- Man, L.L.; Liu, F.; Wang, Y.J.; Song, H.H.; Xu, H.B.; Zhu, Z.W.; Zhang, Q.; Wang, Y.J. The HMGB1 signaling pathway activates the inflammatory response in Schwann cells. Neural Regen. Res. 2015, 10, 1706–1712. [Google Scholar]
- Salvador, B.; Arranz, A.; Francisco, S.; Cordoba, L.; Punzon, C.; Llamas, M.A.; Fresno, M. Modulation of endothelial function by Toll like receptors. Pharmacol. Res. 2016, 108, 46–56. [Google Scholar] [CrossRef]
- Jiang, T.; Jiang, D.; Zhang, L.; Ding, M.; Zhou, H. Anagliptin ameliorates high glucose-induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol. Immunol. 2019, 107, 54–60. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Z.Z.; Zhang, Y.M.; Kong, L.; Xiao, W.F.; Ma, T.F.; Liu, Y.W. Thrombin/PAR-1 activation induces endothelial damages via NLRP1 inflammasome in gestational diabetes. Biochem. Pharmacol. 2020, 175, 113849. [Google Scholar] [CrossRef] [PubMed]
- Misumi, Y.; Ando, Y.; Goncalves, N.P.; Saraiva, M.J. Fibroblasts endocytose and degrade transthyretin aggregates in transthyretin-related amyloidosis. Lab. Investig. 2013, 93, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Fella, E.; Sokratous, K.; Papacharalambous, R.; Kyriacou, K.; Phillips, J.; Sanderson, S.; Panayiotou, E.; Kyriakides, T. Pharmacological Stimulation of Phagocytosis Enhances Amyloid Plaque Clearance; Evidence from a Transgenic Mouse Model of ATTR Neuropathy. Front. Mol. Neurosci. 2017, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Panayiotou, E.; Fella, E.; Papacharalambous, R.; Malas, S.; Saraiva, M.J.; Kyriakides, T. C1q ablation exacerbates amyloid deposition: A study in a transgenic mouse model of ATTRV30M amyloid neuropathy. PLoS ONE 2017, 12, e0175767. [Google Scholar] [CrossRef]
- Garcia-Garcia, M.A.R.; Argilés, À.; Gouin-Charnet, A.; Durfort, M.; Garcia-Valero, J.; Mourad, G. Impaired lysosomal processing of beta2-microglobulin by infiltrating macrophages in dialysis amyloidosis. Kidney Int. 1999, 55, 899–906. [Google Scholar] [CrossRef]
- Okoshi, T.; Yamaguchi, I.; Ozawa, D.; Hasegawa, K.; Naiki, H. Endocytosed 2-Microglobulin Amyloid Fibrils Induce Necrosis and Apoptosis of Rabbit Synovial Fibroblasts by Disrupting Endosomal/Lysosomal Membranes: A Novel Mechanism on the Cytotoxicity of Amyloid Fibrils. PLoS ONE 2015, 10, e0139330. [Google Scholar] [CrossRef]
- Suenaga, G.; Ikeda, T.; Komohara, Y.; Takamatsu, K.; Kakuma, T.; Tasaki, M.; Misumi, Y.; Ueda, M.; Ito, T.; Senju, S.; et al. Involvement of Macrophages in the Pathogenesis of Familial Amyloid Polyneuropathy and Efficacy of Human iPS Cell-Derived Macrophages in Its Treatment. PLoS ONE 2016, 11, e0163944. [Google Scholar] [CrossRef]
- Jabaut, J.; Ather, J.L.; Taracanova, A.; Poynter, M.E.; Ckless, K. Mitochondria-targeted drugs enhance Nlrp3 inflammasome-dependent IL-1beta secretion in association with alterations in cellular redox and energy status. Free Radic. Biol. Med. 2013, 60, 233–245. [Google Scholar] [CrossRef]
- Niemi, K.; Teirila, L.; Lappalainen, J.; Rajamaki, K.; Baumann, M.H.; Oorni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef]
- Westwell-Roper, C.; Nackiewicz, D.; Dan, M.; Ehses, J.A. Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes. Immunol. Cell Biol. 2014, 92, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.J.; Tang, L.; Zhao, X.P.; Xu, J.M.; Xiao, Y.; Li, H. Infiltration of Blood-Derived Macrophages Contributes to the Development of Diabetic Neuropathy. J. Immunol. Res. 2019, 2019, 7597382. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Vazquez, P.A.; Grosick, R.L.; Moracho-Vilrriales, C.; Ward, E.; Threatt, T.; Romero-Sandoval, E.A. Cytokine production capabilities of human primary monocyte-derived macrophages from patients with diabetes mellitus type 2 with and without diabetic peripheral neuropathy. J. Pain Res. 2019, 12, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Couture, R.; Hong, Y. Activated microglia in the spinal cord underlies diabetic neuropathic pain. Eur. J. Pharmacol. 2014, 728, 59–66. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Ledo, J.H.; Barbosa, G.; Sobrinho, M.; Diniz, L.; Fonseca, A.C.; Gomes, F.; Romao, L.; Lima, F.R.; Palhano, F.L.; et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis. Cell Death Dis. 2013, 4, e789. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, H.; Taheri, S.; Mondy, W.; Bonilha, L.; Magwood, G.S.; Lackland, D.; Adams, R.J.; Kindy, M.S. Serum Amyloid A-Mediated Inflammasome Activation of Microglial Cells in Cerebral Ischemia. J. Neurosci. 2019, 39, 9465–9476. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154, S10–S28. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, H.; Hamel, K.A.; Morvan, M.G.; Yu, S.; Leff, J.; Guan, Z.; Braz, J.M.; Basbaum, A.I. Dorsal root ganglion macrophages contribute to both the initiation and persistence of neuropathic pain. Nat. Commun. 2020, 11, 264. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M. Microglia in the spinal cord and neuropathic pain. J. Diabetes Investig. 2016, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Rajchgot, T.; Thomas, S.C.; Wang, J.C.; Ahmadi, M.; Balood, M.; Crosson, T.; Dias, J.P.; Couture, R.; Claing, A.; Talbot, S. Neurons and Microglia; A Sickly-Sweet Duo in Diabetic Pain Neuropathy. Front. Neurosci. 2019, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Y.; Cao, L.; Xiong, R.; Zhang, B.; Wu, L.; Zhao, Z.; Chen, S.D. Curcumin could reduce the monomer of TTR with Tyr114Cys mutation via autophagy in cell model of familial amyloid polyneuropathy. Drug Des. Dev. Ther. 2014, 8, 2121–2128. [Google Scholar]
- Teixeira, C.A.; Almeida Mdo, R.; Saraiva, M.J. Impairment of autophagy by TTR V30M aggregates: In vivo reversal by TUDCA and curcumin. Clin. Sci. Lond. 2016, 130, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, M.; Ju, Y.; Li, A.; Sun, X. Autophagy dysfunction in neuropathic pain. Neuropeptides 2019, 75, 41–48. [Google Scholar] [CrossRef]
- Yin, Y.; Yi, M.H.; Kim, D.W. Impaired Autophagy of GABAergic Interneurons in Neuropathic Pain. Pain Res. Manag. 2018, 2018, 9185368. [Google Scholar] [CrossRef]
- Haidar, M.; Timmerman, V. Autophagy as an Emerging Common Pathomechanism in Inherited Peripheral Neuropathies. Front. Mol. Neurosci. 2017, 10, 143. [Google Scholar] [CrossRef]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.M.; Oh, S.H.; Jin, S.M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient beta cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef]
- Osorio, J. Diabetes: Protective role of autophagy in pancreatic beta cells. Nat. Rev. Endocrinol. 2014, 10, 575. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic beta cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.; Park, K.; Lee, M.S. beta-cell autophagy: Mechanism and role in beta-cell dysfunction. Mol. Metab. 2019, 27, S92–S103. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Resende, R.; Moreira, P.I.; Pereira, C.M. Alzheimer’s disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol. 2015, 34, 261–273. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.M.; Hernandez, N.; Sproul, A.A.; Yu, W.H. Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 2019, 697, 49–58. [Google Scholar] [CrossRef]
- Plaza-Zabala, A.; Sierra-Torre, V.; Sierra, A. Autophagy and Microglia: Novel Partners in Neurodegeneration and Aging. Int. J. Mol. Sci. 2017, 18, 598. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, R.; Bokil, N.J.; Sweet, M.J. Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: A ticking time bomb in ageing. Ageing Res. Rev. 2015, 24, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.; Martins, R.; Bharadwaj, P. Autophagy Modulation as a Treatment of Amyloid Diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [PubMed]
- Said, G. Familial amyloid polyneuropathy: Mechanisms leading to nerve degeneration. Amyloid 2003, 10, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Lozeron, P.; Mariani, L.L.; Dodet, P.; Beaudonnet, G.; Theaudin, M.; Adam, C.; Arnulf, B.; Adams, D. Transthyretin amyloid polyneuropathies mimicking a demyelinating polyneuropathy. Neurology 2018, 91, e143–e152. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Fei, Y.; Su, W.; Chen, G. Emerging Role of Schwann Cells in Neuropathic Pain: Receptors, Glial Mediators and Myelination. Front. Cell Neurosci. 2019, 13, 116. [Google Scholar] [CrossRef]
- Sousa, M.M.; Cardoso, I.; Fernandes, R.; Guimaraes, A.; Saraiva, M.J. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: Evidence for toxicity of nonfibrillar aggregates. Am. J. Pathol. 2001, 159, 1993–2000. [Google Scholar] [CrossRef]
- Murakami, T.; Sango, K.; Watabe, K.; Niimi, N.; Takaku, S.; Li, Z.; Yamamura, K.; Sunada, Y. Schwann cells contribute to neurodegeneration in transthyretin amyloidosis. J. Neurochem. 2015, 134, 66–74. [Google Scholar] [CrossRef]
- Jang, S.Y.; Shin, Y.K.; Lee, H.Y.; Park, J.Y.; Suh, D.J.; Kim, J.K.; Bae, Y.S.; Park, H.T. Local production of serum amyloid a is implicated in the induction of macrophage chemoattractants in Schwann cells during wallerian degeneration of peripheral nerves. Glia 2012, 60, 1619–1628. [Google Scholar] [CrossRef]
- Koike, H.; Katsuno, M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines 2019, 7, 11. [Google Scholar] [CrossRef]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, M.; Mourad, G.; Durfort, M.; Garcia-Valero, J.; Argiles, A. Vascular involvement and cell damage in experimental AA and clinical beta(2)-microglobulin amyloidosis. Nephrol. Dial. Transplant. 2002, 17, 1450–1456. [Google Scholar] [CrossRef] [PubMed]
- Belmokhtar, K.; Robert, T.; Ortillon, J.; Braconnier, A.; Vuiblet, V.; Boulagnon-Rombi, C.; Diebold, M.D.; Pietrement, C.; Schmidt, A.M.; Rieu, P.; et al. Signaling of Serum Amyloid A Through Receptor for Advanced Glycation End Products as a Possible Mechanism for Uremia-Related Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, J.; Wang, S. Serum Amyloid A Induces a Vascular Smooth Muscle Cell Phenotype Switch through the p38 MAPK Signaling Pathway. Biomed. Res. Int. 2017, 2017, 4941379. [Google Scholar] [CrossRef] [PubMed]
- Nishida, E.; Aino, M.; Kobayashi, S.I.; Okada, K.; Ohno, T.; Kikuchi, T.; Hayashi, J.I.; Yamamoto, G.; Hasegawa, Y.; Mitani, A. Serum Amyloid A Promotes E-Selectin Expression via Toll-Like Receptor 2 in Human Aortic Endothelial Cells. Mediators Inflamm. 2016, 2016, 7150509. [Google Scholar] [CrossRef] [PubMed]
- Chami, B.; Barrie, N.; Cai, X.; Wang, X.; Paul, M.; Morton-Chandra, R.; Sharland, A.; Dennis, J.M.; Freedman, S.B.; Witting, P.K. Serum amyloid A receptor blockade and incorporation into high-density lipoprotein modulates its pro-inflammatory and pro-thrombotic activities on vascular endothelial cells. Int. J. Mol. Sci. 2015, 16, 11101–11124. [Google Scholar] [CrossRef]
- Brissova, M.; Shostak, A.; Fligner, C.L.; Revetta, F.L.; Washington, M.K.; Powers, A.C.; Hull, R.L. Human Islets Have Fewer Blood Vessels than Mouse Islets and the Density of Islet Vascular Structures Is Increased in Type 2 Diabetes. J. Histochem. Cytochem. 2015, 63, 637–645. [Google Scholar] [CrossRef]
- Hayden, M.R.; Karuparthi, P.R.; Habibi, J.; Lastra, G.; Patel, K.; Wasekar, C.; Manrique, C.M.; Ozerdem, U.; Stas, S.; Sowers, J.R. Ultrastructure of islet microcirculation, pericytes and the islet exocrine interface in the HIP rat model of diabetes. Exp. Biol. Med. Maywood 2008, 233, 1109–1123. [Google Scholar] [CrossRef]
- Clark, A.; Nilsson, M.R. Islet amyloid: A complication of islet dysfunction or an aetiological factor in Type 2 diabetes? Diabetologia 2004, 47, 157–169. [Google Scholar] [CrossRef]
- Ly, H.; Verma, N.; Wu, F.; Liu, M.; Saatman, K.E.; Nelson, P.T.; Slevin, J.T.; Goldstein, L.B.; Biessels, G.J.; Despa, F. Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann. Neurol. 2017, 82, 208–222. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L. Increased permeability across the blood-nerve barrier of albumin glycated in vitro and in vivo from patients with diabetic polyneuropathy. Proc. Natl. Acad. Sci. USA 1992, 89, 2218–2222. [Google Scholar] [CrossRef] [PubMed]
- Mizisin, A.P.; Weerasuriya, A. Homeostatic regulation of the endoneurial microenvironment during development, aging and in response to trauma, disease and toxic insult. Acta Neuropathol. 2011, 121, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Richner, M.; Ferreira, N.; Dudele, A.; Jensen, T.S.; Vaegter, C.B.; Goncalves, N.P. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front. Neurosci. 2018, 12, 1038. [Google Scholar] [CrossRef]
- Bengoechea, T.G.; Chen, Z.; O’Leary, D.A.; Masliah, E.; Lee, K.F. p75 reduces beta-amyloid-induced sympathetic innervation deficits in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2009, 106, 7870–7875. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of amyloid protein aggregation. The β-strands in the amyloid-forming protein are indicated as red arrows and α-helices as blue spheres. In the native conformation, β-strands of the monomeric protein (if present) are not aligned and “shielded”, which prevents intermolecular aggregation. A partially unfolded or misfolded molecule can form different kinds of intermolecular aggregates. Amyloid oligomers are relatively small, compact structures that may be composed of antiparallel β-strands or contain α-helical conformations. Protofibrils and mature amyloid fibrils are formed via β-strand stacking, forming extended networks of β-sheets with a characteristic cross-beta structure. Mature fibrils consist of a few identical fibrillar “subunits”. Smaller aggregates (oligomers and protofibrils) are mostly cytotoxic, whereas extracellular, fibrillar amyloid deposits can also impair tissue and organ function by impairing blood supply to the cells (see text for references).
Figure 1.
Schematic representation of amyloid protein aggregation. The β-strands in the amyloid-forming protein are indicated as red arrows and α-helices as blue spheres. In the native conformation, β-strands of the monomeric protein (if present) are not aligned and “shielded”, which prevents intermolecular aggregation. A partially unfolded or misfolded molecule can form different kinds of intermolecular aggregates. Amyloid oligomers are relatively small, compact structures that may be composed of antiparallel β-strands or contain α-helical conformations. Protofibrils and mature amyloid fibrils are formed via β-strand stacking, forming extended networks of β-sheets with a characteristic cross-beta structure. Mature fibrils consist of a few identical fibrillar “subunits”. Smaller aggregates (oligomers and protofibrils) are mostly cytotoxic, whereas extracellular, fibrillar amyloid deposits can also impair tissue and organ function by impairing blood supply to the cells (see text for references).

Figure 2.
Schematic representation of the ultrastructure of a peripheral sensory nerve, with locations where amyloid or amyloid protein aggregates have been demonstrated indicated by a red bar (based on references [73,74,75,76,77,78,79,80,81]). DRG = dorsal root ganglion.

Figure 3.
Amyloid deposits in/around small endoneurial blood vessels in the left sural nerve of a 70-year old patient with immunoglobulin light chain amyloidosis. (A) White light microscopy of Congo red stained section, showing pink-stained thickening of vascular walls in a nerve fasciculus. These thickened vascular walls also stained positive for lambda light chains (not shown). (B,C) Enlargement of the framed area of the top panel, (B) viewed with white light, (C) viewed with polarized light, showing green/yellow birefringence of the Congo red positive vascular walls, proving the amyloid nature of these light chain deposits.
Figure 3.
Amyloid deposits in/around small endoneurial blood vessels in the left sural nerve of a 70-year old patient with immunoglobulin light chain amyloidosis. (A) White light microscopy of Congo red stained section, showing pink-stained thickening of vascular walls in a nerve fasciculus. These thickened vascular walls also stained positive for lambda light chains (not shown). (B,C) Enlargement of the framed area of the top panel, (B) viewed with white light, (C) viewed with polarized light, showing green/yellow birefringence of the Congo red positive vascular walls, proving the amyloid nature of these light chain deposits.

Figure 4.
Overview of the cell biological mechanisms that have been implicated in amyloid-protein-induced cellular damage and apoptosis and peripheral neuropathy, both in cells producing an amyloid protein and in other cells affected by amyloid or amyloid protein aggregates. Such “amyloid target cells” include cell types that degrade amyloid and protein aggregates after phagocytosis (macrophages and microglia), as well as Schwann cells (involved in nerve function and integrity) and endothelial cells (involved in microangiopathy).
Figure 4.
Overview of the cell biological mechanisms that have been implicated in amyloid-protein-induced cellular damage and apoptosis and peripheral neuropathy, both in cells producing an amyloid protein and in other cells affected by amyloid or amyloid protein aggregates. Such “amyloid target cells” include cell types that degrade amyloid and protein aggregates after phagocytosis (macrophages and microglia), as well as Schwann cells (involved in nerve function and integrity) and endothelial cells (involved in microangiopathy).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Albariqi, M.M.H.; Engelsman, S.; Eijkelkamp, N.; Höppener, J.W.M. Amyloid Proteins and Peripheral Neuropathy. Cells 2020, 9, 1553. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061553
AMA Style
Albariqi MMH, Engelsman S, Eijkelkamp N, Höppener JWM. Amyloid Proteins and Peripheral Neuropathy. Cells. 2020; 9(6):1553. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061553
Chicago/Turabian StyleAlbariqi, Mohammed M. H., Sjoukje Engelsman, Niels Eijkelkamp, and Jo W. M. Höppener. 2020. "Amyloid Proteins and Peripheral Neuropathy" Cells 9, no. 6: 1553. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9061553
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.