Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Summary Statement
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Constructs and Site-Directed Mutagenesis
2.2. Cell Culture and Transfection
2.3. Knockdown of RAB10, RAB8A, or RAB29 by RNAi
2.4. Immunofluorescence and Laser Confocal Imaging
2.5. Fluorescent EGF Binding and Uptake Assays
2.6. Cell Extracts and Western Blotting
2.7. GST-RILP Pulldown Assays for Determination of Active RAB7
2.8. Statistical Analysis
3. Results
3.1. G2019S LRRK2-Mediated Endolysosomal Trafficking Defects are Rescued by Active RAB10 and Mimicked by Knockdown of RAB10
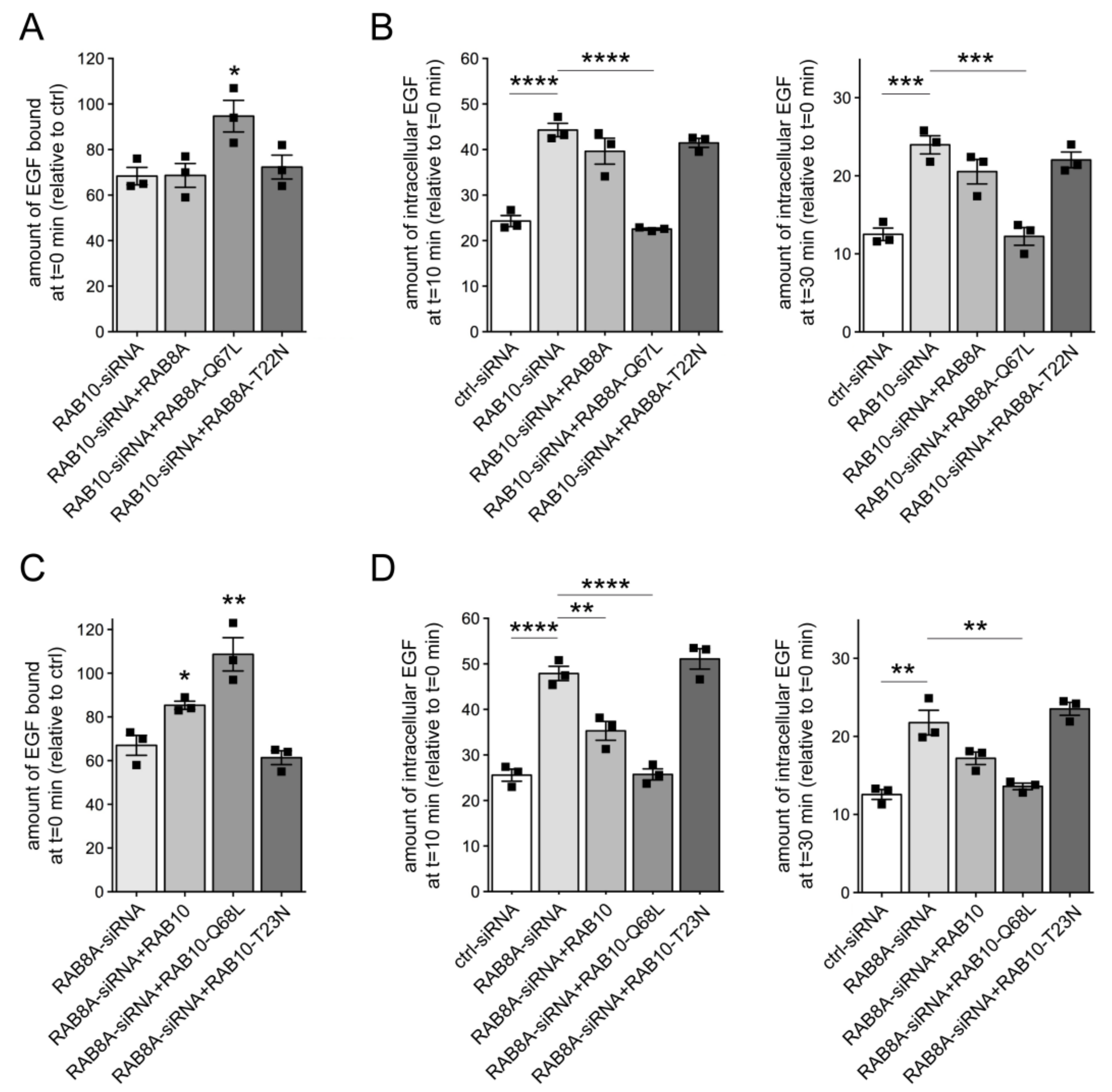
3.2. Knockdown of RAB10 Causes a Decrease in RAB7 Activity and Mistargeting of EGF into an RAB4 Compartment
3.3. G2019S LRRK2-Mediated Endolysosomal Trafficking Defects are Rescued by RAB29 Expression
3.4. RAB29 Expression Rescues the Endolysosomal Trafficking Deficits Mediated by G2019S LRRK2 or Knockdown of Either RAB8A or RAB10
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alessi, D.R.; Sammler, E. LRRK2 kinase in Parkinson’s disease. Science 2018, 360, 36–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.W.; Pei, Z.; Jiang, H.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat. Neurosci. 2006, 9, 1231–1233. [Google Scholar] [CrossRef]
- Jaleel, M.; Nichols, R.J.; Deak, M.; Campbell, D.G.; Gillardon, F.; Knebel, A.; Alessi, D.R. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 2007, 405, 307–317. [Google Scholar] [CrossRef]
- West, A.B.; Moore, D.J.; Choi, C.; Andrabi, S.A.; Li, X.; Dikeman, D.; Biskup, S.; Zhang, Z.; Lim, K.L.; Dawson, V.L.; et al. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 2007, 16, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Luzón-Toro, B.; de la Torre, E.R.; Delgado, A.; Pérez-Tur, J.; Hilfiker, S. Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum. Mol. Genet. 2007, 16, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.D.; Shin, J.-H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.-I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef] [Green Version]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase activity and neuronal toxicity of parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Hatcher, J.; Choi, H.; Alessi, D.; Gray, N. Small-Molecule Inhibitors of LRRK2. Adv Neurobiol. 2017, 14, 241–264. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 2017, 6, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Wandinger-Ness, A.; Zerial, M. Rab Proteins and the Compartmentalization of the Endosomal System. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef]
- Pfeffer, S.R. Rab GTPases: master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell 2017, 28, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Lamber, E.P.; Siedenburg, A.-C.; Barr, F.A. Rab regulation by GEFs and GAPs during membrane traffic. Curr. Opin. Cell Biol. 2019, 59, 34–39. [Google Scholar] [CrossRef]
- Itzen, A.; Goody, R.S. GTPases involved in vesicular trafficking: Structures and mechanisms. Semin. Cell Dev. Biol. 2011, 22, 48–56. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 5, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Madero-Pérez, J.; Fdez, E.; Fernández, B.; Lara Ordóñez, A.J.; Blanca Ramírez, M.; Gómez-Suaga, P.; Waschbüsch, D.; Lobbestael, E.; Baekelandt, V.; Nairn, A.C.; et al. Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol. Neurodegener. 2018, 13, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Rivero-Ríos, P.; Romo-Lozano, M.; Fasiczka, R.; Naaldijk, Y.; Hilfiker, S. LRRK2-Related Parkinson’s Disease Due to Altered Endolysosomal Biology With Variable Lewy Body Pathology: A Hypothesis. Front. Neurosci. 2020, 14, 556. [Google Scholar] [CrossRef]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; MacCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 Interacts with LRRK2 to Modify Intraneuronal Protein Sorting and Parkinson’s Disease Risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beilina, A.; Rudenko, I.N.; Kaganovich, A.; Civiero, L.; Chau, H.; Kalia, S.K.; Kalia, L.V.; Lobbestael, E.; Chia, R.; Ndukwe, K.; et al. Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl. Acad. Sci. USA 2014, 111, 2626–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, H.J.; Kim, H.; Ga, I.; Oh, H.; Ho, D.H.; Kim, J.; Seo, H.; Son, I.; Seol, W. An early endosome regulator, Rab5b, is an LRRK2 kinase substrate. J. Biochem. 2015, 157, 485–495. [Google Scholar] [CrossRef]
- Ito, G.; Katsemonova, K.; Tonelli, F.; Lis, P.; Baptista, M.A.S.; Shpiro, N.; Duddy, G.; Wilson, S.; Ho, P.W.-L.; Ho, S.-L.; et al. Phos-tag analysis of Rab10 phosphorylation by LRRK2: a powerful assay for assessing kinase function and inhibitors. Biochem. J. 2016, 473, 2671–2685. [Google Scholar] [CrossRef] [Green Version]
- Thirstrup, K.; Dächsel, J.C.; Oppermann, F.S.; Williamson, D.S.; Smith, G.P.; Fog, K.; Christensen, K.V. Selective LRRK2 kinase inhibition reduces phosphorylation of endogenous Rab10 and Rab12 in human peripheral mononuclear blood cells. Sci. Rep. 2017, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Beilina, A.; Bonet-Ponce, L.; Kumaran, R.; Kordich, J.J.; Ishida, M.; Mamais, A.; Kaganovich, A.; Saez-Atienzar, S.; Gershlick, D.C.; Roosen, D.A.; et al. The Parkinson’s Disease Protein LRRK2 Interacts with the GARP Complex to Promote Retrograde Transport to the trans-Golgi Network. Cell Rep. 2020, 31, 107614. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Odorizzi, G.; Emr, S.D. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002, 3, 893–905. [Google Scholar] [CrossRef]
- Chi, S.; Cao, H.; Wang, Y.; McNiven, M.A. Recycling of the epidermal growth factor receptor is mediated by a novel form of the clathrin adaptor protein Eps15. J. Biol. Chem. 2011, 286, 35196–35208. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Suaga, P.; Rivero-Ríos, P.; Fdez, E.; Blanca Ramírez, M.; Ferrer, I.; Aiastui, A.; López De Munain, A.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef] [Green Version]
- Rivero-Ríos, P.; Romo-Lozano, M.; Madero-Pérez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The G2019S variant of leucine-rich repeat kinase 2 (LRRK2) alters endolysosomal trafficking by impairing the function of the GTPase RAB8A. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef] [Green Version]
- Fernández, B.; Lara Ordóñez, A.J.; Fdez, E.; Mutez, E.; Comptdaer, T.; Leghay, C.; Kreisler, A.; Simonin, C.; Vandewynckel, L.; Defebvre, L.; et al. Centrosomal cohesion deficits as cellular biomarker in lymphoblastoid cell lines from LRRK2 Parkinson’s disease patients. Biochem. J. 2019, 476, 2797–2813. [Google Scholar] [CrossRef] [Green Version]
- Lis, P.; Burel, S.; Steger, M.; Mann, M.; Brown, F.; Diez, F.; Tonelli, F.; Holton, J.L.; Ho, P.W.; Ho, S.-L.; et al. Development of phospho-specific Rab protein antibodies to monitor in vivo activity of the LRRK2 Parkinson’s disease kinase. Biochem. J. 2018, 475, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Howden, A.J.M.; Sarhan, A.R.; Lis, P.; Ito, G.; Martinez, T.N.; Brockmann, K.; Gasser, T.; Alessi, D.R.; Sammler, E.M. Interrogating Parkinson’s disease LRRK2 kinase pathway activity by assessing Rab10 phosphorylation in human neutrophils. Biochem. J. 2018, 475, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Atashrazm, F.; Hammond, D.; Perera, G.; Bolliger, M.F.; Matar, E.; Halliday, G.M.; Schüle, B.; Lewis, S.J.G.; Nichols, R.J.; Dzamko, N. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov. Disord. 2019, 34, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbey, C.M.; Ahktar, N.; Wang, E.; Chih-Hsiung Cheng, C.; Grant, B.D.; Dunn, E.W. Rab10 Regulates Membrane Transport through Early Endosomes of Polarized Madin-Darby Canine Kidney Cells. Mol. Biol. Cell 2006, 17, 3156–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roland, J.T.; Lapierre, L.A.; Goldenring, J.R. Alternative Splicing in Class V Myosins Determines Association with Rab10. J. Biol. Chem. 2009, 284, 1213–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lara Ordónez, A.J.; Fernández, B.; Fdez, E.; Romo-Lozano, M.; Madero-Pérez, J.; Lobbestael, E.; Baekelandt, V.; Aiastui, A.; López de Munaín, A.; Melrose, H.L.; et al. RAB8, RAB10 and RILPL1 contribute to both LRRK2 kinase–mediated centrosomal cohesion and ciliogenesis deficits. Hum. Mol. Genet. 2019, 28, 3552–3568. [Google Scholar] [CrossRef] [Green Version]
- Etoh, K.; Fukuda, M. Rab10 regulates tubular endosome formation through KIF13A and KIF13B motors. J. Cell Sci. 2019, 132, jcs226977. [Google Scholar] [CrossRef] [Green Version]
- Chua, C.E.L.; Tang, B.L. Rab 10—A traffic controller in multiple cellular pathways and locations. J. Cell. Physiol. 2018, 233, 6483–6494. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Iwano, T.; Kunii, M.; Matsuda, S.; Mizuguchi, R.; Jung, Y.; Hagiwara, H.; Yoshihara, Y.; Yuzaki, M.; Harada, R.; et al. Rab8a and Rab8b are essential for several apical transport pathways but insufficient for ciliogenesis. J. Cell Sci. 2014, 127, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S.; Gerl, M.J.; Ang, A.; Manninen, A.; Keller, P.; Mellman, I.; Simons, K. Rab10 is Involved in Basolateral Transport in Polarized Madin–Darby Canine Kidney Cells. Traffic 2007, 8, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Fukuda, M. Rabin8 regulates neurite outgrowth in both GEF activity-dependent and -independent manners. Mol. Biol. Cell 2016, 27, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Tao, T.; Sun, J.; Peng, Y.; Li, Y.; Wang, P.; Chen, X.; Zhao, W.; Zheng, Y.-Y.; Wei, L.; Wang, W.; et al. Golgi-resident TRIO regulates membrane trafficking during neurite outgrowth. J. Biol. Chem. 2019, 294, 10954–10968. [Google Scholar] [CrossRef] [PubMed]
- Tucci, A.; Nalls, M.A.; Houlden, H.; Revesz, T.; Singleton, A.B.; Wood, N.W.; Hardy, J.; Paisán-Ruiz, C. Genetic variability at the PARK16 locus. Eur. J. Hum. Genet. 2010, 18, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Bar-Shira, A.; Dahary, D.; Mirelman, A.; Kedmi, M.; Gurevich, T.; Giladi, N.; Orr-Urtreger, A. Association of Sequence Alterations in the Putative Promoter of RAB7L1 With a Reduced Parkinson Disease Risk. Arch. Neurol. 2012, 69, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.-Y.; Chen, Y.-P.; Song, W.; Zhao, B.; Cao, B.; Wei, Q.-Q.; Ou, R.-W.; Yang, Y.; Yuan, L.-X.; Shang, H.-F. An association analysis of the rs1572931 polymorphism of the RAB7L1 gene in Parkinson’s disease, amyotrophic lateral sclerosis and multiple system atrophy in China. Eur. J. Neurol. 2014, 21, 1337–1343. [Google Scholar] [CrossRef]
- Pihlstrøm, L.; Rengmark, A.; Bjørnarå, K.A.; Dizdar, N.; Fardell, C.; Forsgren, L.; Holmberg, B.; Larsen, J.P.; Linder, J.; Nissbrandt, H.; et al. Fine mapping and resequencing of the PARK16 locus in Parkinson’s disease. J. Hum. Genet. 2015, 60, 357–362. [Google Scholar] [CrossRef]
- Khaligh, A.; Goudarzian, M.; Moslem, A.; Mehrtash, A.; Jamshidi, J.; Darvish, H.; Emamalizadeh, B. RAB7L1 promoter polymorphism and risk of Parkinson’s disease; a case-control study. Neurol. Res. 2017, 39, 468–471. [Google Scholar] [CrossRef]
- He, T.; Wang, J.; Wang, X.; Deng, W.; Jiang, H.; Xie, J.; Sun, P. Association between PARK16 and Parkinson’s disease: A meta-analysis. Neurosci. Lett. 2017, 657, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Goudarzian, M.; Khaligh, A.; Fourozan, R.; Jamal Mirmoosavi, S.; Darvish, H.; Safaralizadeh, T.; Emamalizadeh, B. The rs1572931 polymorphism of the RAB7L1 gene promoter is associated with reduced risk of Parkinson’s disease. Neurol. Res. 2015, 37, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Soto-Ortolaza, A.I.; Heckman, M.G.; Labbé, C.; Serie, D.J.; Puschmann, A.; Rayaprolu, S.; Strongosky, A.; Boczarska-Jedynak, M.; Opala, G.; Krygowska-Wajs, A.; et al. GWAS risk factors in Parkinson’s disease: LRRK2 coding variation and genetic interaction with PARK16. Am. J. Neurodegener. Dis. 2013, 2, 287–299. [Google Scholar] [PubMed]
- Liu, Z.; Bryant, N.; Kumaran, R.; Beilina, A.; Abeliovich, A.; Cookson, M.R.; West, A.B. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet. 2018, 27, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Madero-Pérez, J.; Fernández, B.; Lara Ordóñez, A.J.; Fdez, E.; Lobbestael, E.; Baekelandt, V.; Hilfiker, S. RAB7L1-Mediated Relocalization of LRRK2 to the Golgi Complex Causes Centrosomal Deficits via RAB8A. Front. Mol. Neurosci. 2018, 11, 417. [Google Scholar] [CrossRef] [Green Version]
- Purlyte, E.; Dhekne, H.S.; Sarhan, A.R.; Gomez, R.; Lis, P.; Wightman, M.; Martinez, T.N.; Tonelli, F.; Pfeffer, S.R.; Alessi, D.R. Rab29 activation of the Parkinson’s disease-associated LRRK2 kinase. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- Gomez, R.C.; Wawro, P.; Lis, P.; Alessi, D.R.; Pfeffer, S.R. Membrane association but not identity is required for LRRK2 activation and phosphorylation of Rab GTPases. J. Cell Biol. 2019, 218, 4157–4170. [Google Scholar] [CrossRef] [Green Version]
- Civiero, L.; Vancraenenbroeck, R.; Belluzzi, E.; Beilina, A.; Lobbestael, E.; Reyniers, L.; Gao, F.; Micetic, I.; de Maeyer, M.; Bubacco, L.; et al. Biochemical Characterization of Highly Purified Leucine-Rich Repeat Kinases 1 and 2 Demonstrates Formation of Homodimers. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Peralta, E.R.; Martin, B.C.; Edinger, A.L. Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence. J. Biol. Chem. 2010, 285, 16814–16821. [Google Scholar] [CrossRef] [Green Version]
- Ceresa, B.P.; Bahr, S.J. Rab7 activity affects epidermal growth factor:epidermal growth factor receptor degradation by regulating endocytic trafficking from the late endosome. J. Biol. Chem. 2006, 281, 1099–1106. [Google Scholar] [CrossRef] [Green Version]
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 Regulates Late Endocytic Trafficking Downstream of Multivesicular Body Biogenesis and Cargo Sequestration. J. Biol. Chem. 2009, 284, 12110–12124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassu, M.; Grazia, M.; Giudice, D.; Sanna, S.; Taymans, J.M.; Morari, M.; Brugnoli, A.; Frassineti, M.; Masala, A.; Esposito, S.; et al. Role of LRRK2 in the regulation of dopamine receptor trafficking. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Heaton, G.R.; Landeck, N.; Mamais, A.; Nalls, M.A.; Nixon-Abell, J.; Kumaran, R.; Beilina, A.; Pellegrini, L.; Li, Y.; Harvey, K.; et al. Sequential screening nominates the Parkinson’s disease associated kinase LRRK2 as a regulator of Clathrin-mediated endocytosis. Neurobiol. Dis. 2020, 141, 104948. [Google Scholar] [CrossRef]
- McCaffrey, M.W.; Bielli, A.; Cantalupo, G.; Mora, S.; Roberti, V.; Santillo, M.; Drummond, F.; Bucci, C. Rab4 affects both recycling and degradative endosomal trafficking. FEBS Lett. 2001, 495, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.C.; Hendrick, J.; Eisler, S.A.; Schmid, S.; Hausser, A.; Olayioye, M.A. The Rho-specific GAP protein DLC3 coordinates endocytic membrane trafficking. J. Cell Sci. 2015, 128, 1386–1399. [Google Scholar] [CrossRef] [Green Version]
- Goueli, B.S.; Powell, M.B.; Finger, E.C.; Pfeffer, S.R. TBC1D16 is a Rab4A GTPase activating protein that regulates receptor recycling and EGF receptor signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 15787–15792. [Google Scholar] [CrossRef] [Green Version]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.R. Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol. 2013, 25, 414–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, F.A. Review series: Rab GTPases and membrane identity: causal or inconsequential? J. Cell Biol. 2013, 202, 191–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langemeyer, L.; Perz, A.; Kümmel, D.; Ungermann, C. A guanine nucleotide exchange factor (GEF) limits Rab GTPase-driven membrane fusion. J. Biol. Chem. 2018, 293, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langemeyer, L.; Borchers, A.-C.; Herrmann, E.; Füllbrunn, N.; Han, Y.; Perz, A.; Auffarth, K.; Kümmel, D.; Ungermann, C. A conserved and regulated mechanism drives endosomal Rab transition. Elife 2020, 9, e56090. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, T.; Inoue, K.; D’Agati, V.D.; Fujimoto, T.; Eguchi, T.; Saha, S.; Wolozin, B.; Iwatsubo, T.; Abeliovich, A. LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep. 2016, 6, 29945. [Google Scholar] [CrossRef]
- Onnis, A.; Finetti, F.; Patrussi, L.; Gottardo, M.; Cassioli, C.; Spanò, S.; Baldari, C.T. The small GTPase Rab29 is a common regulator of immune synapse assembly and ciliogenesis. Cell Death Differ. 2015, 22, 1687–1699. [Google Scholar] [CrossRef] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivero-Ríos, P.; Romo-Lozano, M.; Fernández, B.; Fdez, E.; Hilfiker, S. Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations. Cells 2020, 9, 1719. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9071719
Rivero-Ríos P, Romo-Lozano M, Fernández B, Fdez E, Hilfiker S. Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations. Cells. 2020; 9(7):1719. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9071719
Chicago/Turabian StyleRivero-Ríos, Pilar, Maria Romo-Lozano, Belén Fernández, Elena Fdez, and Sabine Hilfiker. 2020. "Distinct Roles for RAB10 and RAB29 in Pathogenic LRRK2-Mediated Endolysosomal Trafficking Alterations" Cells 9, no. 7: 1719. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9071719