Fyn Tyrosine Kinase Elicits Amyloid Precursor Protein Tyr682 Phosphorylation in Neurons from Alzheimer’s Disease Patients

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Neural Progenitors
2.2. Immunoprecipitation (IP) Assay and Western Blot (WB) Analysis
2.3. Inhibitors
2.4. ELISA
2.5. Materials
2.6. Statistical Analysis
3. Results
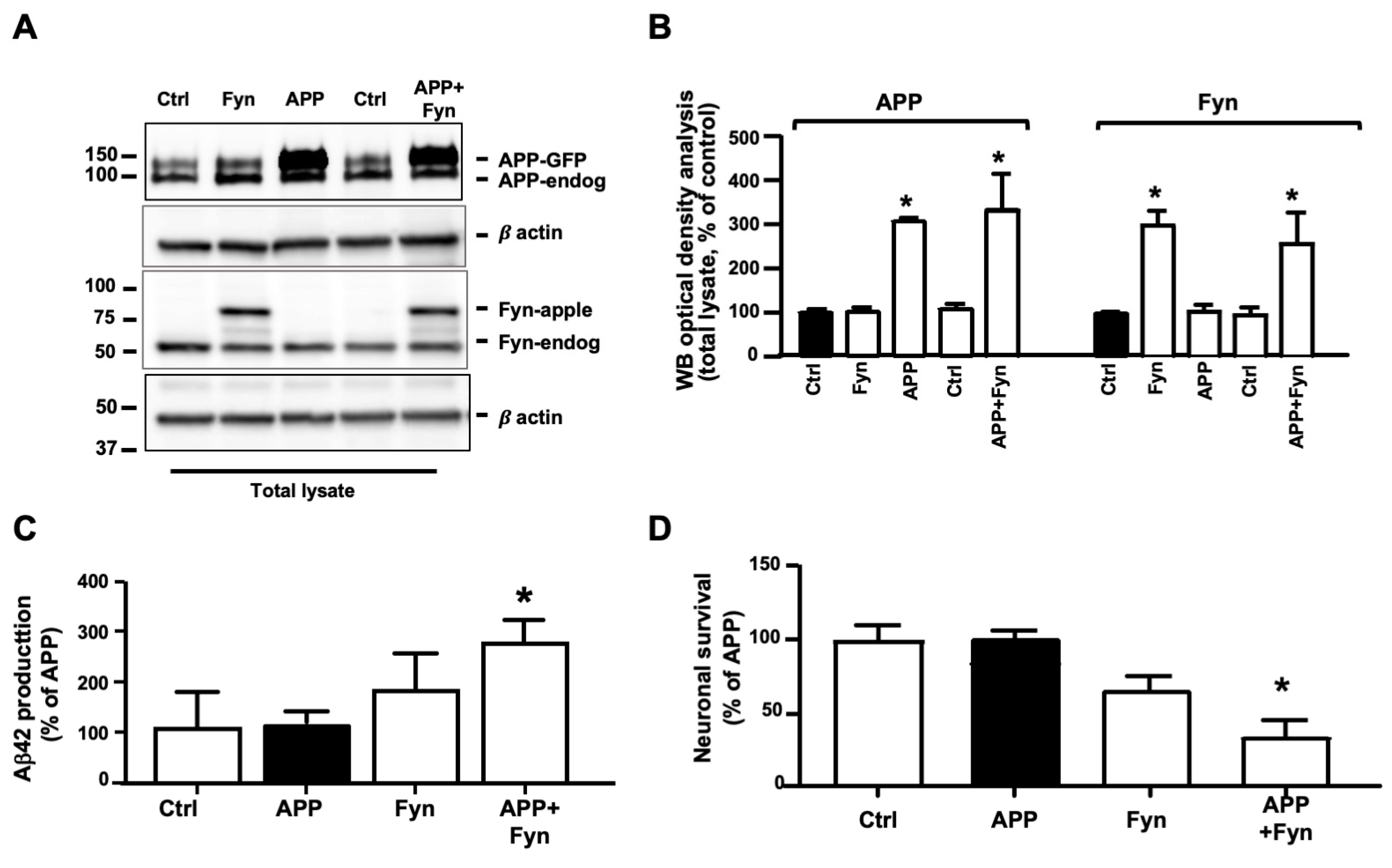
3.1. Fyn Overexpression and Overactivation Promoted APP Amyloidogenic Processing and Neuronal Death in Human Neurons
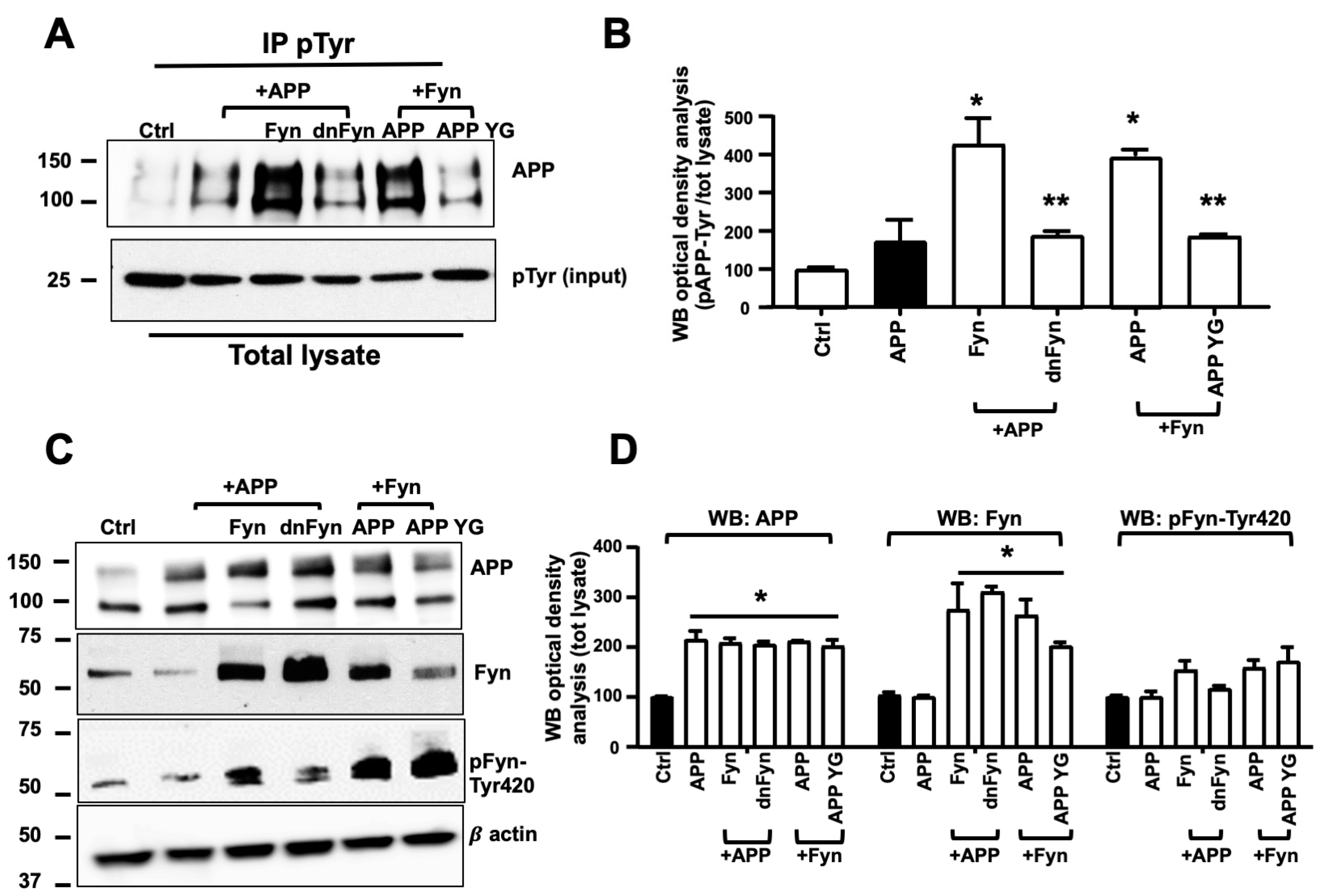
3.2. Fyn Overactivation Failed in Phosphorylating APP When Tyr682 Was Replaced by Gly in Human Neurons
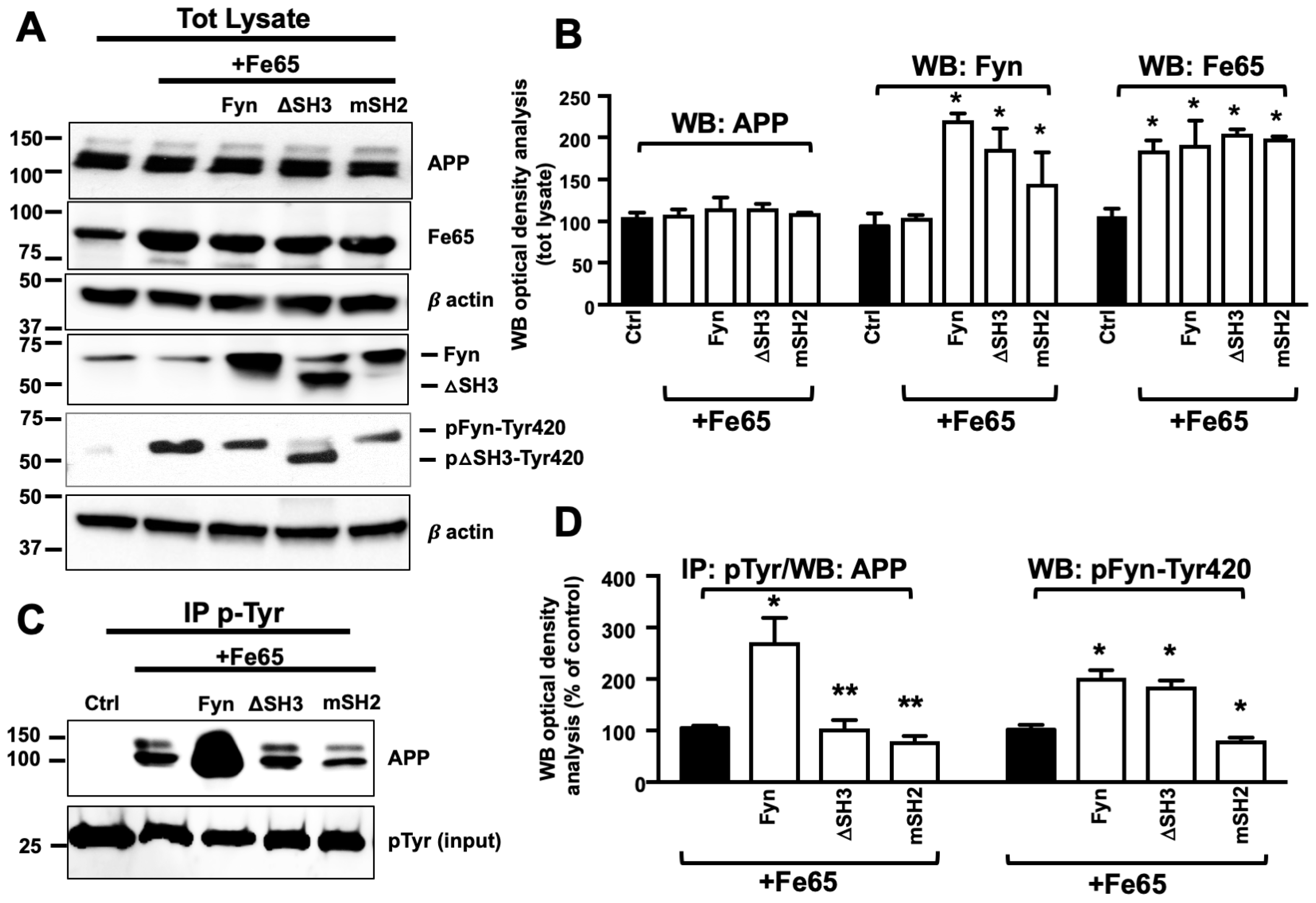
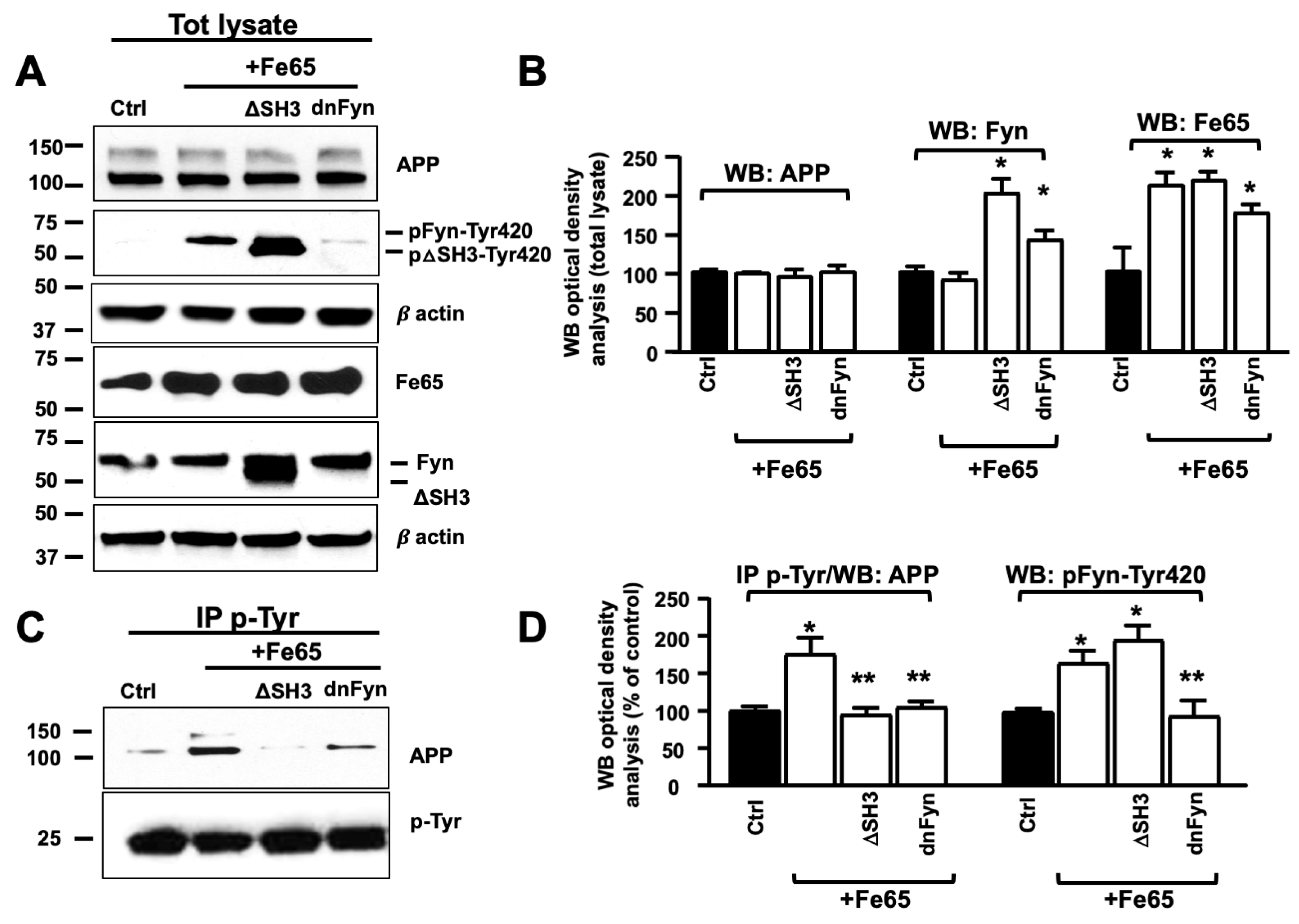
3.3. Fe65 Promoted Fyn Mediated APP Tyr682 Phosphorylation in Human Neurons
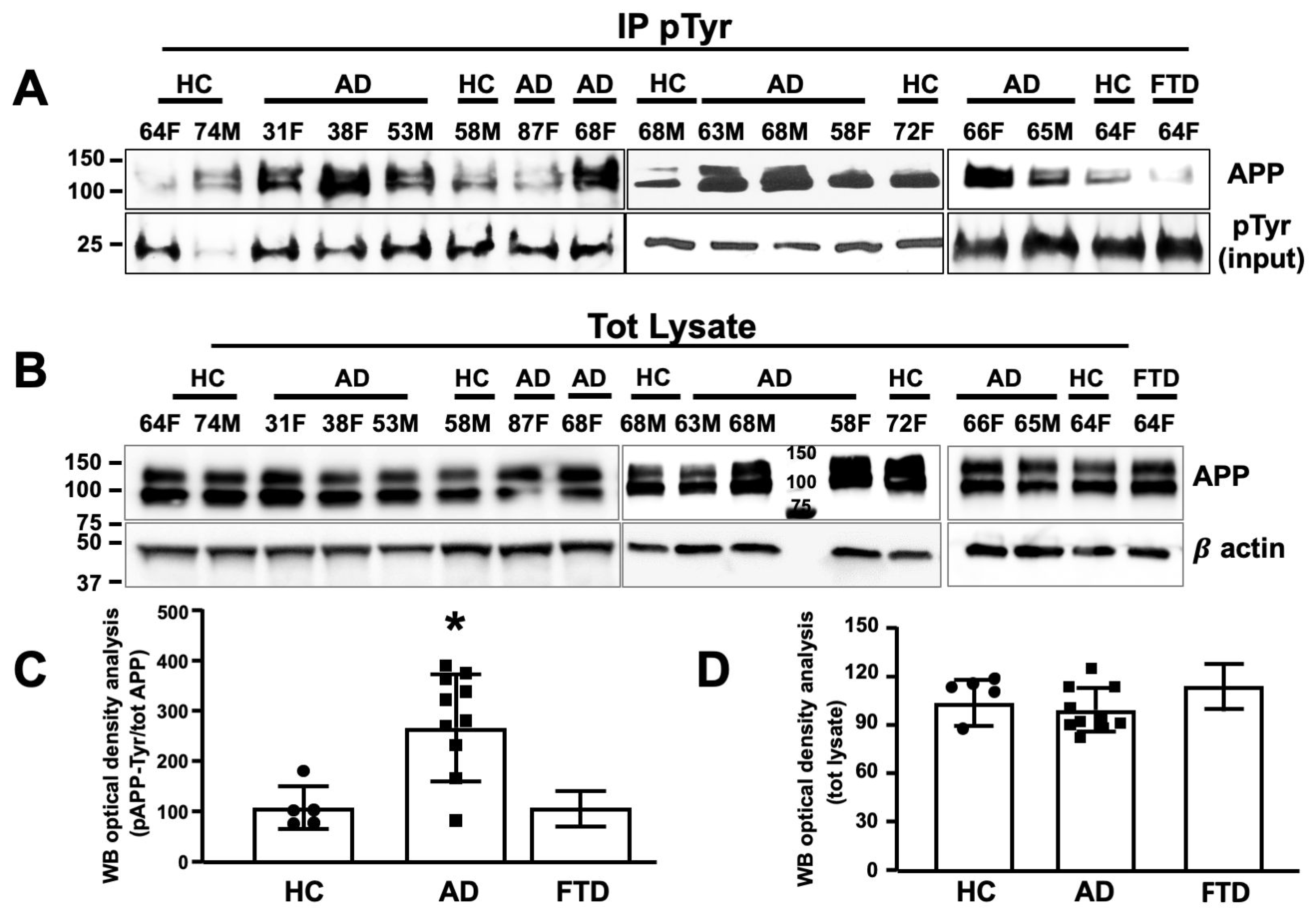
3.4. APP Tyr Phosphorylation Was Higher in Neurons from AD Patients as Compared with Neurons from Healthy Volunteers
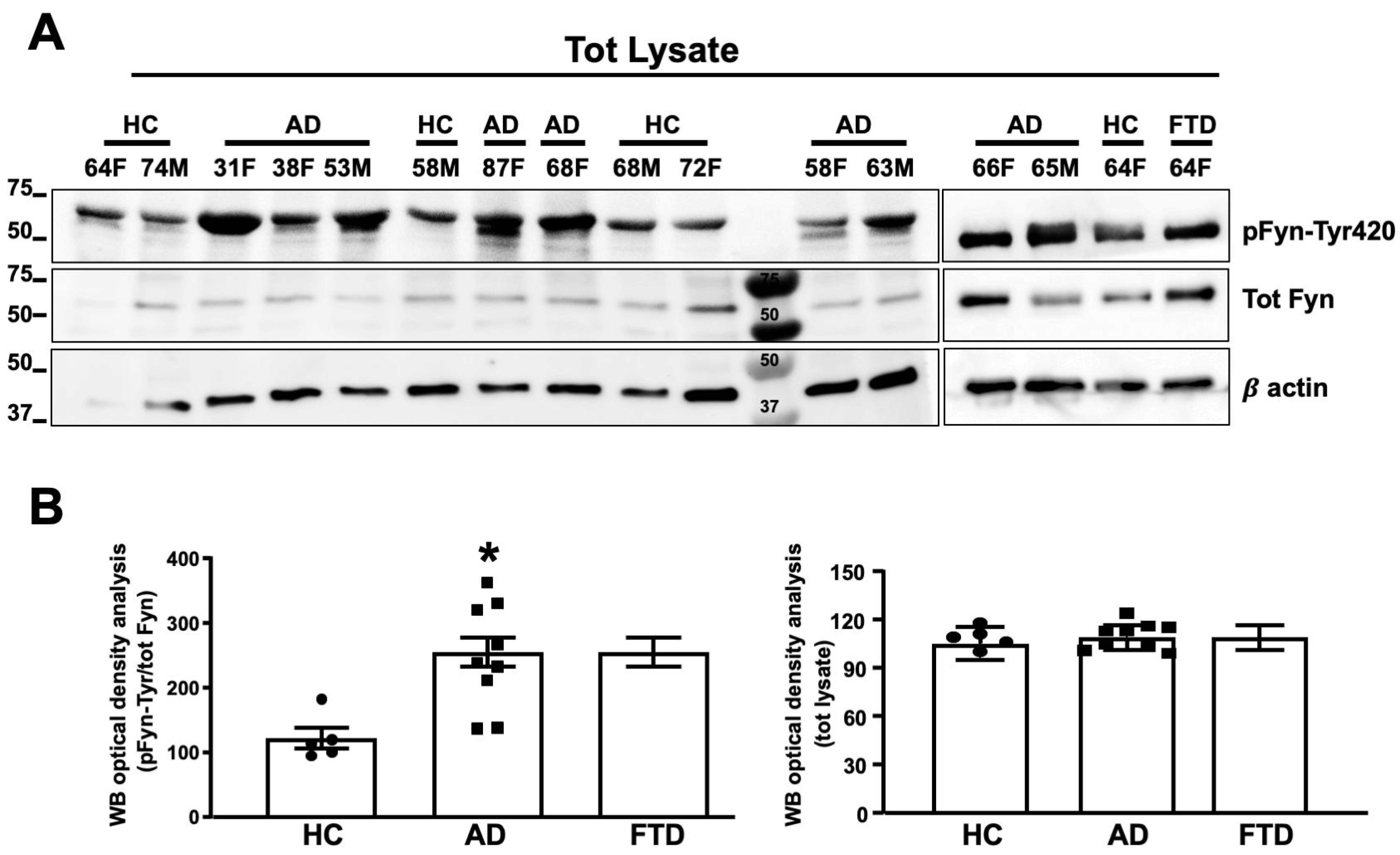
3.5. Fyn TK Activity Was Increased in Neurons from AD Patients as Compared to Healthy Volunteers
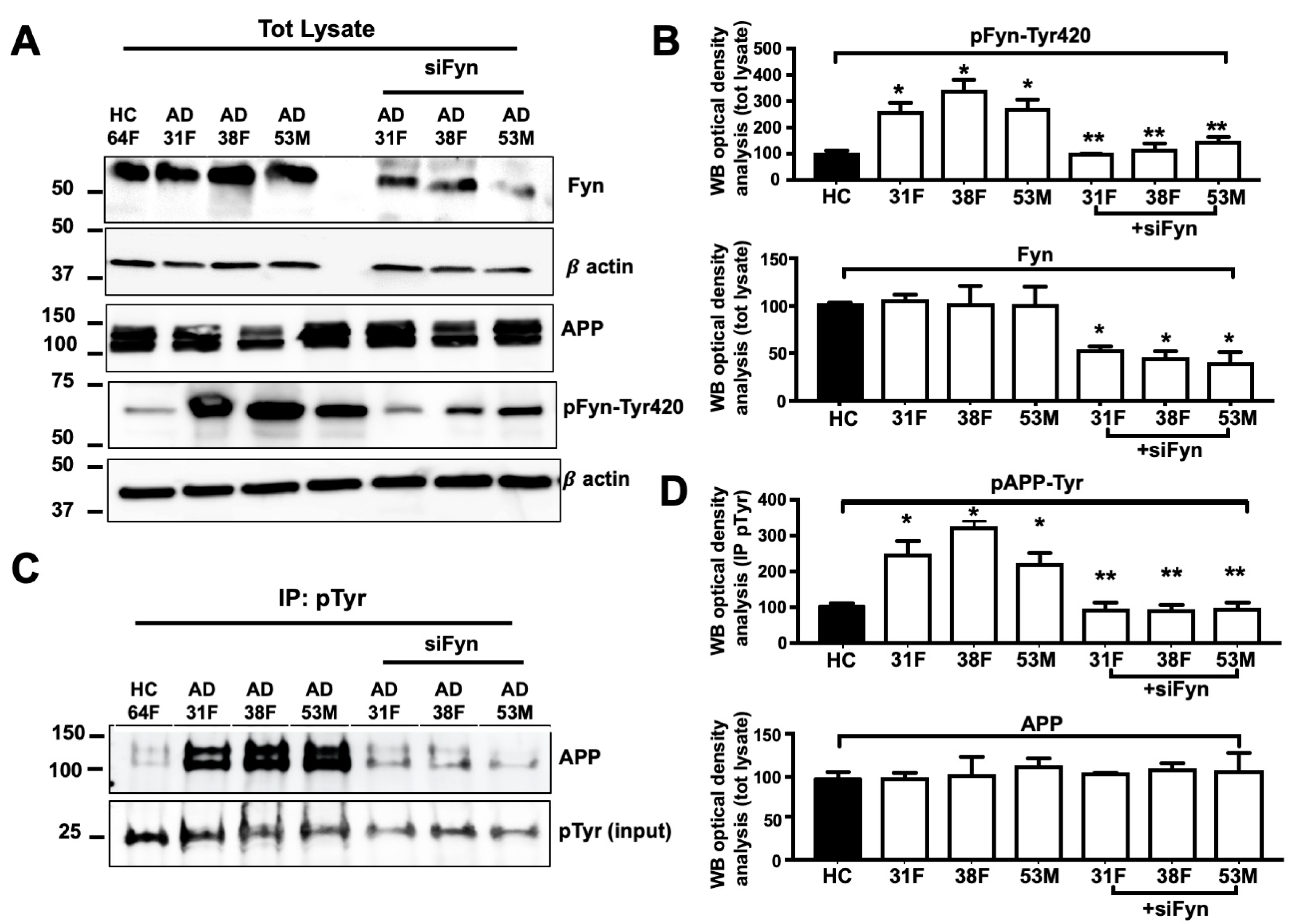
3.6. APP Tyr Phosphorylation Was Decreased in Neurons in Which Fyn Expression Levels Were Knocked Down
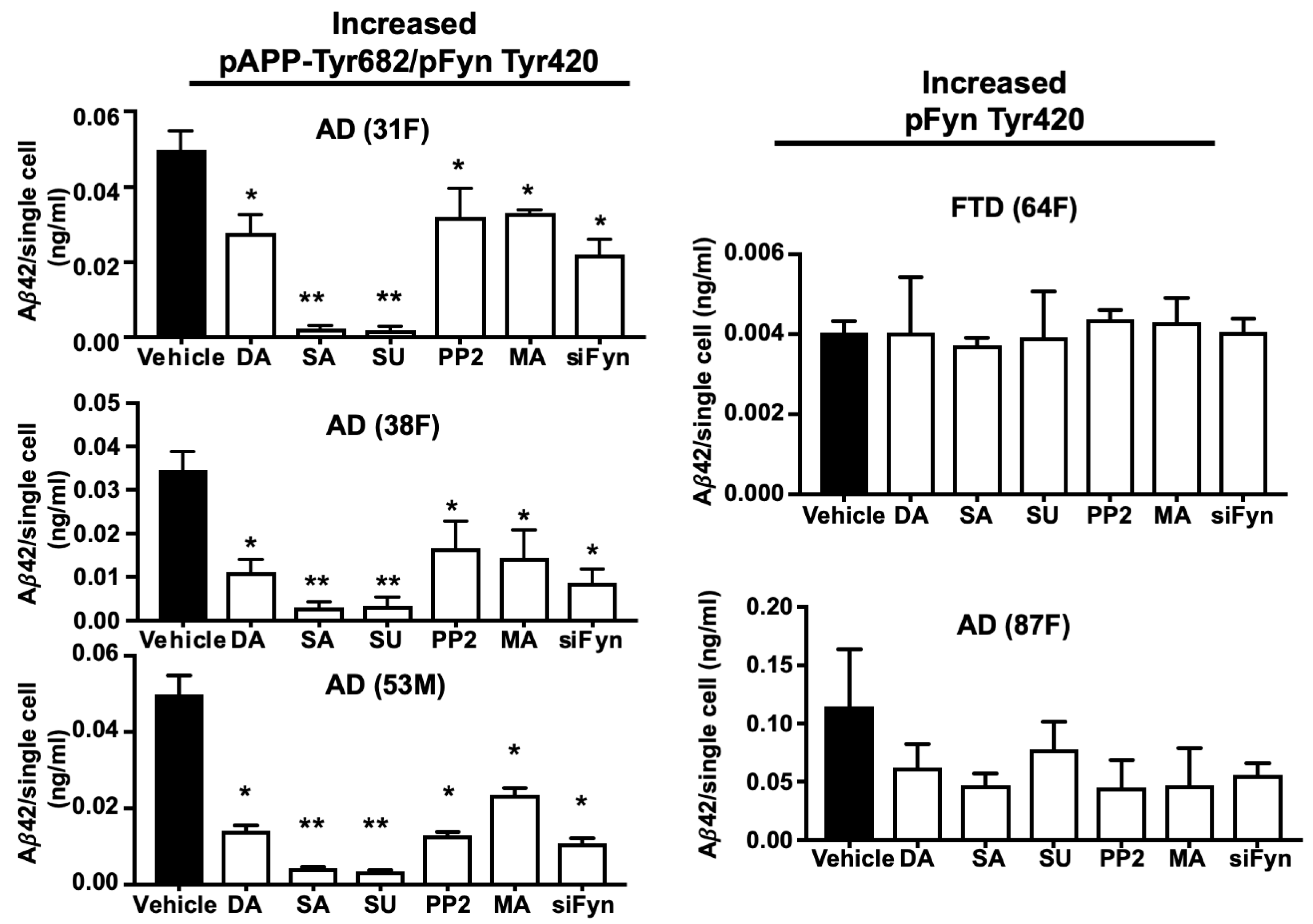
3.7. TKI Exposure Inhibited Aβ42 Release in Neurons from AD Patients with High APP Tyr Phosphorylation Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Walsh, D.M.; Selkoe, D.J. Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The path forward in Alzheimer’s disease therapeutics: Reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020. [Google Scholar] [CrossRef]
- Armstrong, R.A. A critical analysis of the ‘amyloid cascade hypothesis’. Folia Neuropathol. 2014, 52, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, J.L.; Christopeit, T.; Villaverde, M.C.; Gossas, T.; Otero, J.M.; Nyström, S.; Baraznenok, V.; Lindström, E.; Danielson, U.H.; Sussman, F. Effect of the protonation state of the titratable residues on the inhibitor affinity to BACE-1. Biochemistry 2010, 49, 7255–7263. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matrone, C.; Iannuzzi, F.; Annunziato, L. The Y682ENPTY687 motif of APP: Progress and insights toward a targeted therapy for Alzheimer’s disease patients. Ageing Res. Rev. 2019, 52, 120–128. [Google Scholar] [CrossRef]
- King, G.D.; Scott Turner, R. Adaptor protein interactions: Modulators of amyloid precursor protein metabolism and Alzheimer’s disease risk? Exp. Neurol. 2004, 185, 208–219. [Google Scholar] [CrossRef]
- Klevanski, M.; Herrmann, U.; Weyer, S.W.; Fol, R.; Cartier, N.; Wolfer, D.P.; Caldwell, J.H.; Korte, M.; Müller, U.C. The APP Intracellular Domain Is Required for Normal Synaptic Morphology, Synaptic Plasticity, and Hippocampus-Dependent Behavior. J. Neurosci. 2015, 35, 16018–16033. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Basso, E.; Matrone, C. NGF and APP Interplay: Focus on YENPTY Motif of Amyloid Precursor Protein and Y682 Residue. Cell Biol. Res. Ther. 2013, 2. [Google Scholar] [CrossRef]
- Tamayev, R.; Zhou, D.; D’Adamio, L. The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol. Neurodegener. 2009, 4, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimberly, W.T.; Zheng, J.B.; Guénette, S.Y.; Selkoe, D.J. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J. Biol. Chem. 2001, 276, 40288–40292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunan, J.; Shearman, M.S.; Checler, F.; Cappai, R.; Evin, G.; Beyreuther, K.; Masters, C.L.; Small, D.H. The C-terminal fragment of the Alzheimer’s disease amyloid protein precursor is degraded by a proteasome-dependent mechanism distinct from gamma-secretase. Eur. J. Biochem. 2001, 268, 5329–5336. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, N.; Bruni, P.; Minopoli, G.; Mosca, R.; Molino, D.; Russo, C.; Schettini, G.; Sudol, M.; Russo, T. The beta-amyloid precursor protein APP is tyrosine-phosphorylated in cells expressing a constitutively active form of the Abl protoncogene. J. Biol. Chem. 2001, 276, 19787–19792. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, A.P.; Wang, Z.; Zheng, H.; D’Adamio, L. A single tyrosine residue in the amyloid precursor protein intracellular domain is essential for developmental function. J. Biol. Chem. 2011, 286, 8717–8721. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, E.T.; Larsen, A.; Zollo, A.; Jørgensen, A.L.; Sanggaard, K.W.; Enghild, J.J.; Matrone, C. New Insights to Clathrin and Adaptor Protein 2 for the Design and Development of Therapeutic Strategies. Int. J. Mol. Sci. 2015, 16, 29446–29453. [Google Scholar] [CrossRef]
- La Rosa, L.R.; Perrone, L.; Nielsen, M.S.; Calissano, P.; Andersen, O.M.; Matrone, C. Y682G Mutation of Amyloid Precursor Protein Promotes Endo-Lysosomal Dysfunction by Disrupting APP-SorLA Interaction. Front. Cell Neurosci. 2015, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C. A new molecular explanation for age-related neurodegeneration: The Tyr682 residue of amyloid precursor protein. Bioessays 2013, 35, 847–852. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Luvisetto, S.; La Rosa, L.R.; Tamayev, R.; Pignataro, A.; Canu, N.; Yang, L.; Barbagallo, A.P.; Biundo, F.; Lombino, F.; et al. Tyr682 in the Abeta-precursor protein intracellular domain regulates synaptic connectivity, cholinergic function, and cognitive performance. Aging Cell 2012, 11, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, E.T.; Iannuzzi, F.; Rasmussen, H.F.; Maier, T.J.; Enghild, J.J.; Jørgensen, A.L.; Matrone, C. An Aberrant Phosphorylation of Amyloid Precursor Protein Tyrosine Regulates Its Trafficking and the Binding to the Clathrin Endocytic Complex in Neural Stem Cells of Alzheimer’s Disease Patients. Front. Mol. Neurosci. 2017, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, J.E.; Marianne, G.J.; Schmidt, M.; Liu, Y.; Li, R.; Henrik, C.; Margarita, M.; Mette, H.; Carmela, M.; Yvonne, B.; et al. Expression of the Alzheimer’s Disease Mutations AβPP695sw and PSEN1M146I in Double-Transgenic Göttingen Minipigs. J. Alzheimer’s Dis. 2016, 53, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Neet, K.; Hunter, T. Vertebrate non-receptor protein-tyrosine kinase families. Genes Cells 1996, 1, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Williamson, R.; Scales, T.; Clark, B.R.; Gibb, G.; Reynolds, C.H.; Kellie, S.; Bird, I.N.; Varndell, I.M.; Sheppard, P.W.; Everall, I.; et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: Involvement of Src family protein kinases. J. Neurosci. 2002, 22, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehm, J. A ‘danse macabre’: Tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. Eur. J. Neurosci. 2013, 37, 1925–1930. [Google Scholar] [CrossRef]
- Chin, J.; Palop, J.J.; Yu, G.Q.; Kojima, N.; Masliah, E.; Mucke, L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 2004, 24, 4692–4697. [Google Scholar] [CrossRef]
- Nakazawa, T.; Komai, S.; Tezuka, T.; Hisatsune, C.; Umemori, H.; Semba, K.; Mishina, M.; Manabe, T.; Yamamoto, T. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001, 276, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Chin, J.; Palop, J.J.; Puoliväli, J.; Massaro, C.; Bien-Ly, N.; Gerstein, H.; Scearce-Levie, K.; Masliah, E.; Mucke, L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2005, 25, 9694–9703. [Google Scholar] [CrossRef] [Green Version]
- Minami, S.S.; Clifford, T.G.; Hoe, H.S.; Matsuoka, Y.; Rebeck, G.W. Fyn knock-down increases Aβ, decreases phospho-tau, and worsens spatial learning in 3× Tg-AD mice. Neurobiol. Aging 2012, 33, 825-e15. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, A.C.; Salazar, S.V.; Haas, L.T.; Yang, J.; Kostylev, M.A.; Jeng, A.T.; Robinson, S.A.; Gunther, E.C.; van Dyck, C.H.; Nygaard, H.B.; et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef]
- Lesort, M.; Jope, R.S.; Johnson, G.V. Insulin transiently increases tau phosphorylation: Involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 1999, 72, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111 Pt 21, 3167–3177. [Google Scholar]
- Zollo, A.; Allen, Z.; Rasmussen, H.F.; Iannuzzi, F.; Shi, Y.; Larsen, A.; Maier, T.J.; Matrone, C. Sortilin-Related Receptor Expression in Human Neural Stem Cells Derived from Alzheimer’s Disease Patients Carrying the APOE Epsilon 4 Allele. Neural Plast 2017, 2017, 1892612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Belrose, J.; Trepanier, C.H.; Lei, G.; Jackson, M.F.; MacDonald, J.F. Fyn, a potential target for Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 243–252. [Google Scholar] [CrossRef]
- Nygaard, H.B.; van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Schettini, G.; Govoni, S.; Racchi, M.; Rodriguez, G. Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: Signal transduction and/or transcriptional role--relevance for Alzheimer pathology. J. Neurochem. 2010, 115, 1299–1308. [Google Scholar] [CrossRef]
- McLoughlin, D.M.; Miller, C.C. The FE65 proteins and Alzheimer’s disease. J. Neurosci. Res. 2008, 86, 744–754. [Google Scholar] [CrossRef]
- Zhou, D.; Zambrano, N.; Russo, T.; D’Adamio, L. Phosphorylation of a tyrosine in the amyloid-beta protein precursor intracellular domain inhibits Fe65 binding and signaling. J. Alzheimer’s Dis. 2009, 16, 301–307. [Google Scholar] [CrossRef]
- Koistinen, N.A.; Bacanu, S.; Iverfeldt, K. Phosphorylation of Fe65 amyloid precursor protein-binding protein in response to neuronal differentiation. Neurosci. Lett. 2016, 613, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Akiyama, M.; Araki, Y.; Sumioka, A.; Shiono, M.; Taru, H.; Nakaya, T.; Yamamoto, T.; Suzuki, T. Intracellular trafficking of the amyloid beta-protein precursor (APP) regulated by novel function of X11-like. PLoS ONE 2011, 6, e22108. [Google Scholar] [CrossRef] [PubMed]
- Minopoli, G.; Gargiulo, A.; Parisi, S.; Russo, T. Fe65 matters: New light on an old molecule. IUBMB Life 2012, 64, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, H.; Glotzbach, A.; Kolbe, K.; Leonhardt, G.; Loosse, C.; Muller, T. Small things matter: Implications of APP intracellular domain AICD nuclear signaling in the progression and pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2017, 156, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Feilen, L.P.; Haubrich, K.; Strecker, P.; Probst, S.; Eggert, S.; Stier, G.; Sinning, I.; Konietzko, U.; Kins, S.; Simon, B.; et al. Fe65-PTB2 Dimerization Mimics Fe65-APP Interaction. Front. Mol. Neurosci. 2017, 10, 140. [Google Scholar] [CrossRef]
- Guenette, S.; Strecker, P.; Kins, S. APP Protein Family Signaling at the Synapse: Insights from Intracellular APP-Binding Proteins. Front. Mol. Neurosci. 2017, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Ermekova, K.S.; Zambrano, N.; Linn, H.; Minopoli, G.; Gertler, F.; Russo, T.; Sudol, M. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J. Biol. Chem. 1997, 272, 32869–32877. [Google Scholar] [CrossRef] [Green Version]
- Ermekova, K.S.; Chang, A.; Zambrano, N.; de Candia, P.; Russo, T.; Sudol, M. Proteins implicated in Alzheimer disease. The role of FE65, a new adapter which binds to beta-amyloid precursor protein. Adv. Exp. Med. Biol. 1998, 446, 161–180. [Google Scholar]
- Müller, T.; Meyer, H.E.; Egensperger, R.; Marcus, K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics-relevance for Alzheimer’s disease. Prog. Neurobiol. 2008, 85, 393–406. [Google Scholar] [CrossRef]
- Müller, U.C.; Zheng, H. Physiological functions of APP family proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, W.N.; Cheung, H.N.; Li, W.; Lau, K.F. FE65: Roles beyond amyloid precursor protein processing. Cell Mol. Biol. Lett. 2015, 20, 66–87. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Tesco, G.; Jeong, W.J.; Lindsley, L.; Eckman, E.A.; Eckman, C.B.; Tanzi, R.E.; Guenette, S.Y. Generation of the beta-amyloid peptide and the amyloid precursor protein C-terminal fragment gamma are potentiated by FE65L1. J. Biol. Chem. 2003, 278, 51100–51107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, S.N.; Desiderio, S. SH2 domains of the protein-tyrosine kinases Blk, Lyn, and Fyn(T) bind distinct sets of phosphoproteins from B lymphocytes. J. Biol. Chem. 1993, 268, 22557–22565. [Google Scholar]
- Shirazi, S.K.; Wood, J.G. The protein tyrosine kinase, fyn, in Alzheimer’s disease pathology. Neuroreport 1993, 4, 435–437. [Google Scholar] [CrossRef]
- Noble, M.E.; Musacchio, A.; Saraste, M.; Courtneidge, S.A.; Wierenga, R.K. Crystal structure of the SH3 domain in human Fyn; comparison of the three-dimensional structures of SH3 domains in tyrosine kinases and spectrin. EMBO J. 1993, 12, 2617–2624. [Google Scholar] [CrossRef]
- Swope, S.L.; Huganir, R.L. Binding of the nicotinic acetylcholine receptor to SH2 domains of Fyn and Fyk protein tyrosine kinases. J. Biol. Chem. 1994, 269, 29817–29824. [Google Scholar]
- Weng, Z.; Thomas, S.M.; Rickles, R.J.; Taylor, J.A.; Brauer, A.W.; Seidel-Dugan, C.; Michael, W.M.; Dreyfuss, G.; Brugge, J.S. Identification of Src, Fyn, and Lyn SH3-binding proteins: Implications for a function of SH3 domains. Mol. Cell Biol. 1994, 14, 4509–4521. [Google Scholar] [CrossRef] [Green Version]
- Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. [Google Scholar] [CrossRef]
- Perkinton, M.S.; Standen, C.L.; Lau, K.F.; Kesavapany, S.; Byers, H.L.; Ward, M.; McLoughlin, D.M.; Miller, C.C. The c-Abl tyrosine kinase phosphorylates the Fe65 adaptor protein to stimulate Fe65/amyloid precursor protein nuclear signaling. J. Biol. Chem. 2004, 279, 22084–22091. [Google Scholar] [CrossRef] [Green Version]
- Russo, C.; Dolcini, V.; Salis, S.; Venezia, V.; Violani, E.; Carlo, P.; Zambrano, N.; Russo, T.; Schettini, G. Signal transduction through tyrosine-phosphorylated carboxy-terminal fragments of APP via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer’s disease brain. Ann. N. Y. Acad. Sci. 2002, 973, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Matrone, C.; Marolda, R.; Ciafre, S.; Ciotti, M.T.; Mercanti, D.; Calissano, P. Tyrosine kinase nerve growth factor receptor switches from prosurvival to proapoptotic activity via Abeta-mediated phosphorylation. Proc. Natl. Acad. Sci. USA 2009, 106, 11358–11363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acevedo, K.M.; Opazo, C.M.; Norrish, D.; Challis, L.M.; Li, Q.X.; White, A.R.; Bush, A.I.; Camakaris, J. Phosphorylation of amyloid precursor protein at threonine 668 is essential for its copper-responsive trafficking in SH-SY5Y neuroblastoma cells. J. Biol. Chem. 2014, 289, 11007–11019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, R.G.; Soriano, S.; Hayes, J.D.; Ostaszewski, B.; Xia, W.; Selkoe, D.J.; Chen, X.; Stokin, G.B.; Koo, E.H. Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J. Biol. Chem. 1999, 274, 18851–18856. [Google Scholar] [CrossRef] [Green Version]
- Yáñez, M.J.; Belbin, O.; Estrada, L.D.; Leal, N.; Contreras, P.S.; Lleó, A.; Burgos, P.V.; Zanlungo, S.; Alvarez, A.R. c-Abl links APP-BACE1 interaction promoting APP amyloidogenic processing in Niemann-Pick type C disease. Biochim. Biophys. Acta 2016, 1862, 2158–2167. [Google Scholar] [CrossRef]
- Matrone, C.; Barbagallo, A.P.; La Rosa, L.R.; Florenzano, F.; Ciotti, M.T.; Mercanti, D.; Chao, M.V.; Calissano, P.; D’Adamio, L. APP is phosphorylated by TrkA and regulates NGF/TrkA signaling. J. Neurosci. 2011, 31, 11756–11761. [Google Scholar] [CrossRef] [Green Version]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef]
- Netzer, W.J.; Bettayeb, K.; Sinha, S.C.; Flajolet, M.; Greengard, P.; Bustos, V. Gleevec shifts APP processing from a β-cleavage to a nonamyloidogenic cleavage. Proc. Natl. Acad. Sci. USA 2017, 114, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimer’s Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Abushouk, A.I.; Negida, A.; Elshenawy, R.A.; Zein, H.; Hammad, A.M.; Menshawy, A.; Mohamed, W.M.Y. C-Abl Inhibition; A Novel Therapeutic Target for Parkinson’s Disease. CNS Neurol. Disord. Drug Targets 2018, 17, 14–21. [Google Scholar] [CrossRef]
- Nygaard, H.B.; Wagner, A.F.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; Strittmatter, K.A.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P.; et al. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyonaga, T.; Smith, L.M.; Finnema, S.J.; Gallezot, J.D.; Naganawa, M.; Bini, J.; Mulnix, T.; Cai, Z.; Ropchan, J.; Huang, Y.; et al. In vivo synaptic density imaging with (11)C-UCB-J detects treatment effects of saracatinib (AZD0530) in a mouse model of Alzheimer’s disease. J. Nucl. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Radzimanowski, J.; Simon, B.; Sattler, M.; Beyreuther, K.; Sinning, I.; Wild, K. Structure of the intracellular domain of the amyloid precursor protein in complex with Fe65-PTB2. EMBO Rep. 2008, 9, 1134–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neural Stem Cells | |||||
| Sampling Age | Gender | ApoE Genotype | AD-Related Mutation | Source (#) | |
| Healthy Controls (HC) | 64 | F | ------ | Ax0019 | |
| 74 | M | E2/E2 | Ax0018 | ||
| Cord blood | ----- | ------ | Ax0015 | ||
| AD Patients (AD) | 38 | F | E3/E3 | PS1 L286V | Ax112 |
| 53 | M | ------ | PS1 M146L | Ax113 | |
| 31 | F | E3/E4 | PS1 A246E | Ax114 | |
| 87 | F | E4/E4 | ------ | Ax111 | |
| Frontotemporal Dementia (FTD) | 64 | F | ------ | MAPT P301L | Ax324 |
| IPSC | |||||
| Sampling Age | Gender | ApoE4 carrier | CSF Aβ Presence | Source (#) | |
| Healthy Controls (HC) | 58 | M | CW70073 | ||
| 72 (diabetic) | F | CW50068 | |||
| 68 | M | CW50028 | |||
| AD Patients (AD) | 68 | F | Yes | Yes | CW50025 |
| 68 | M | Yes | Yes | CW50082 | |
| 58 | F | Yes | Yes | CW50018 | |
| 63 | M | No | Yes | CW50024 | |
| 66 | F | ---- | Yes | CW50069 | |
| 65 | M | ---- | Yes | CW50126 | |
| IC50 of Tyrosine Kinase Inhibitors Active on Fyn | |
|---|---|
| Dasatinib (DA) | 0.8 nM |
| Saracatinib (SA) | 2.7 nM |
| SU6656 (SU) | 130 nM |
| PP2 | 4 nM / 5 nM |
| Masitinib (MA) | 10 nM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannuzzi, F.; Sirabella, R.; Canu, N.; Maier, T.J.; Annunziato, L.; Matrone, C. Fyn Tyrosine Kinase Elicits Amyloid Precursor Protein Tyr682 Phosphorylation in Neurons from Alzheimer’s Disease Patients. Cells 2020, 9, 1807. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9081807
Iannuzzi F, Sirabella R, Canu N, Maier TJ, Annunziato L, Matrone C. Fyn Tyrosine Kinase Elicits Amyloid Precursor Protein Tyr682 Phosphorylation in Neurons from Alzheimer’s Disease Patients. Cells. 2020; 9(8):1807. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9081807
Chicago/Turabian StyleIannuzzi, Filomena, Rossana Sirabella, Nadia Canu, Thorsten J. Maier, Lucio Annunziato, and Carmela Matrone. 2020. "Fyn Tyrosine Kinase Elicits Amyloid Precursor Protein Tyr682 Phosphorylation in Neurons from Alzheimer’s Disease Patients" Cells 9, no. 8: 1807. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9081807