Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK) Enhances Activation of STAT3/NLRC4 Inflammasome Signaling Axis through PKCδ in Astrocytes: Implications for Parkinson’s Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Chemicals and Biological Reagents
2.2. Human Astrocyte (U373) Cell Culture and Treatment
2.3. LUHMES Human Dopaminergic Neuronal Cell Culture and Treatment
2.4. Primary Astrocyte Culture
2.5. Animal Treatment
2.6. MPTP Mouse Model
2.7. Human Sample Analysis
2.8. Immunohistochemistry and Immunofluorescence
2.9. Live Cell Staining
2.10. Mitochondrial Fragmentation Analysis
2.11. Multiplex Cytokine Luminex Immunoassays
2.12. Intracellular Reactive Oxygen Species (iROS) and MitoSox Assays
2.13. Astroglial Nitric Oxide Detection
2.14. MTS Assay
2.15. ELISA
2.16. Real-Time Quantitative Reverse Transcription PCR (qRT-PCR)
2.17. Western Blotting
2.18. siRNA Transfection
3. Data Analysis
4. Results
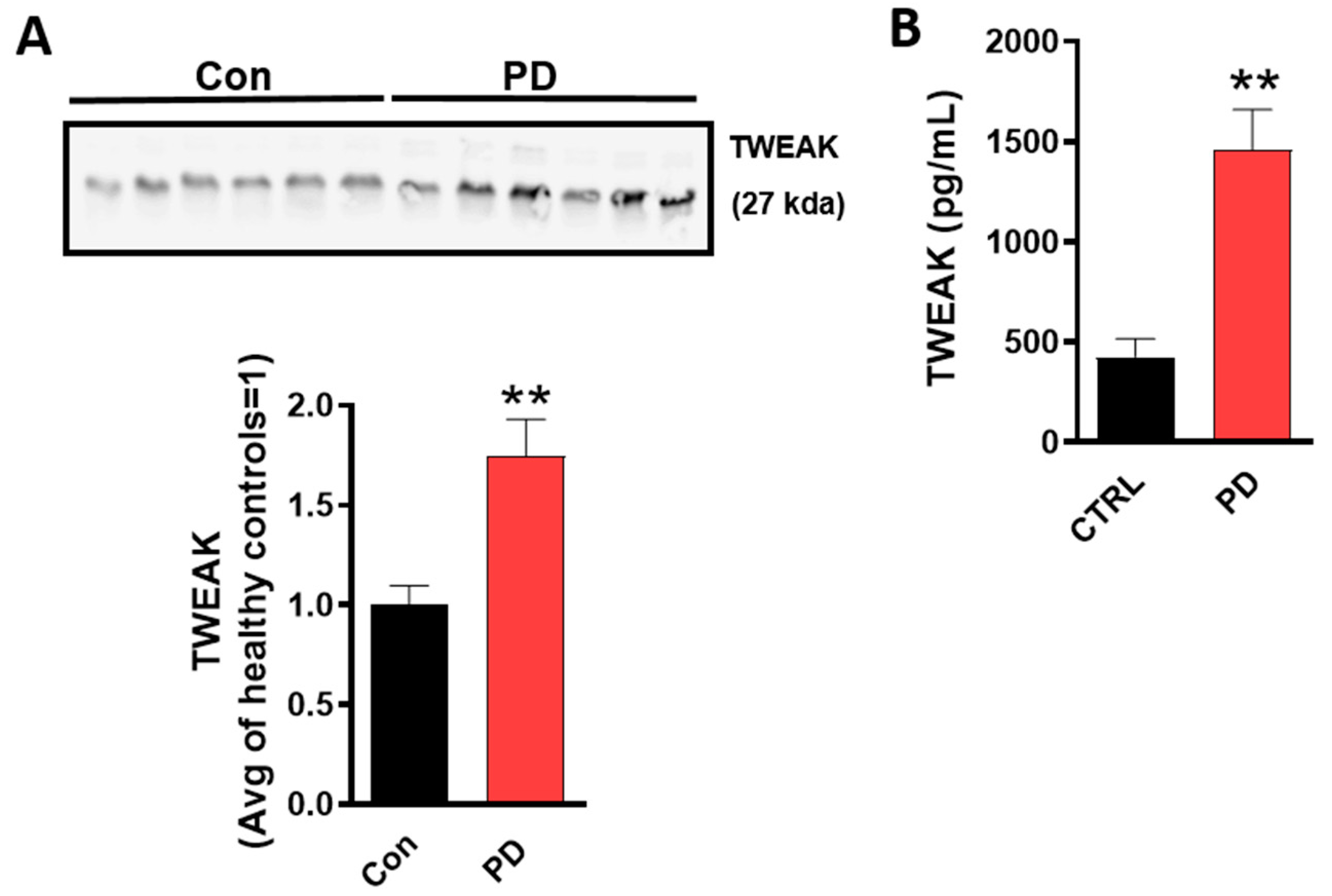
4.1. TWEAK Expression is Elevated in the Serum of Human PD Patients
4.2. Oxidative Stress Mechanisms and Mitochondrial Impairment as well as PKCδ and STAT3 Activation Are Augmented in TWEAK-Treated U373 Astrocyte Cells
4.3. TWEAK Induces NLRC4 Inflammasome Activation in U373 Astrocyte Cells.
4.4. TWEAK Stimulation of Primary Astrocytes Is Associated with the Activation of PKCδ, NLRC4 and STAT3, as well as the Generation of Proinflammatory Cytokines
4.5. TWEAK-Induced ROS Generation, NLRC4 Inflammasome Activation and STAT3 Activation Are Regulated by PKCδ in U373 Cells
4.6. Pharmacological Inhibition of Mitochondrially-Derived ROS via Mito-Apocynin Suppresses Mitochondrial Impairment with a Concomitant Reduction in PKCδ and NLRC4 Inflammasome Activation as well as STAT-3 Activation in Astrocytes
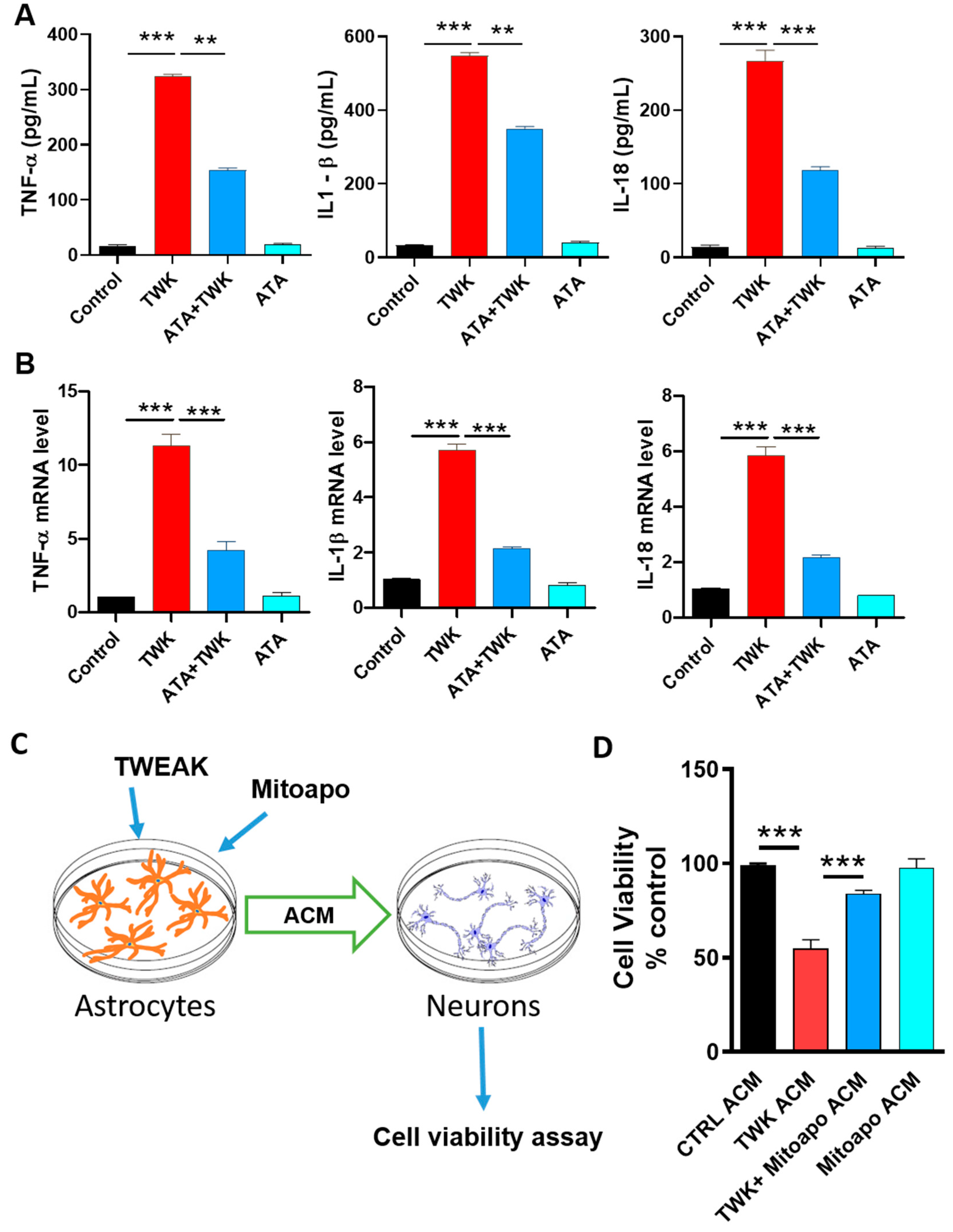
4.7. Inhibition of TWEAK-Fn14 Signaling Attenuated TWEAK-Induced Proinflammatory Cytokine Generation
4.8. Inhibiting Astrocytic Mitochondrial Oxidative Stress Suppressed TWEAK Astrocyte-Conditioned Media (ACM)-Induced Dopaminergic Neurotoxicity
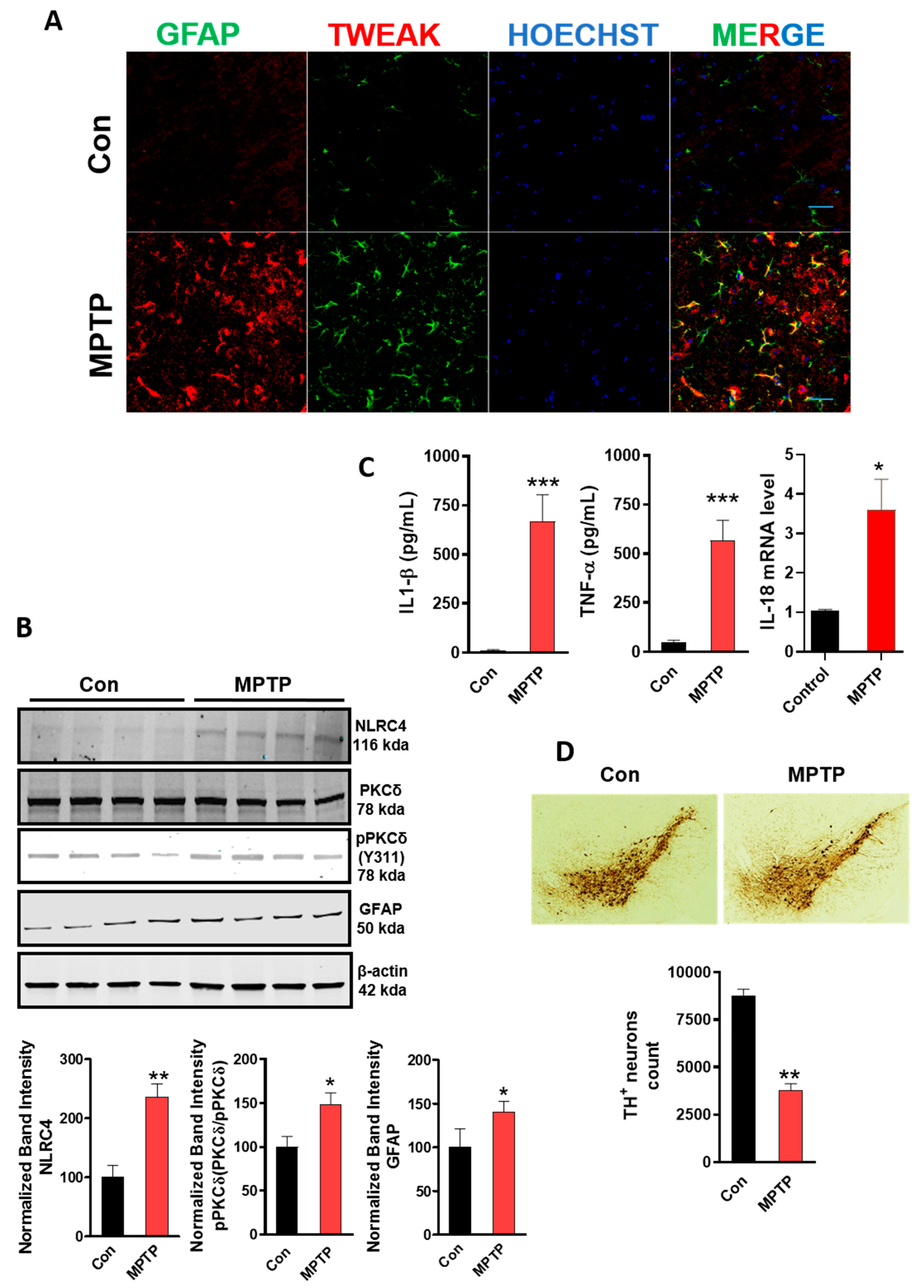
4.9. MPTP-Induced Astrocyte Activation Involves the Upregulation of TWEAK with Concomitant NLRC4 Inflammasome Activation and TH Neuronal Loss
4.10. TWEAK Enhances NLRC4 Inflammasome Activation and Astrogliosis in the Striata
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lotharius, J.; Brundin, P. Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Wolters, E.C.; van der Werf, Y.D.; van den Heuvel, O.A. Parkinson’s disease-related disorders in the impulsive-compulsive spectrum. J. Neurol. 2008, 255, 48. [Google Scholar] [CrossRef] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s Disease and Its Management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Tarale, P.; Sivanesan, S.; Daiwile, A.P.; Stöger, R.; Bafana, A.; Naoghare, P.K.; Parmar, D.; Chakrabarti, T.; Kannan, K. Global DNA methylation profiling of manganese-exposed human neuroblastoma SH-SY5Y cells reveals epigenetic alterations in Parkinson’s disease-associated genes. Arch. Toxicol. 2017, 91, 2629–2641. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.-S.; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Hong, J.S.; Crews, F.T. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia 2013, 61, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.S. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ. Health. Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Whitton, P.S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. Br. J. Pharmacol. 2007, 150, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Nagatsu, T.; Mogi, M.; Ichinose, H.; Togari, A. Cytokines in Parkinson’s disease. J. Neural Transm. Suppl. 2000, 58, 143–151. [Google Scholar]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism. Relat. Disord. 2012, 18 (Suppl. 1), S210–S212. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends. Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.F.; Cardani, S.; Maroli, A.; Vitiello, A.; Soffientini, P.; Crespi, A.; Bram, R.F.; Benfante, R.; Borgese, N. Tail-anchored Protein Insertion in Mammals: FUNCTION AND RECIPROCAL INTERACTIONS OF THE TWO SUBUNITS OF THE TRC40 RECEPTOR. J. Biol. Chem. 2016, 291, 15292–15306. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Cornwell, A.; Li, J.; Peng, S.; Osorio, M.J.; Aalling, N.; Wang, S.; Benraiss, A.; Lou, N.; Goldman, S.A.; et al. SOX9 Is an Astrocyte-Specific Nuclear Marker in the Adult Brain Outside the Neurogenic Regions. J. Neurosci. 2017, 37, 4493–4507. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, J.P.; Kelly, K.A.; VanGilder, R.L.; Sofroniew, M.V.; Miller, D.B. Early activation of STAT3 regulates reactive astrogliosis induced by diverse forms of neurotoxicity. PLoS ONE 2014, 9, e102003. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Xue, T.; Zhang, Z.; Wang, X.; Xu, P.; Zhang, J.; Lei, X.; Li, Y.; Xie, Y.; Wang, L.; et al. Role of signal transducer and activator of transcription-3 in up-regulation of GFAP after epilepsy. Neurochem. Res. 2011, 36, 2208–2215. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflamm. 2009, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Lawana, V.; Singh, N.; Sarkar, S.; Charli, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. Involvement of c-Abl Kinase in Microglial Activation of NLRP3 Inflammasome and Impairment in Autolysosomal System. J. Neuroimmune. Pharmacol. 2017, 12, 624–660. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell. Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [Green Version]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Liu, L.; Chan, C. IPAF inflammasome is involved in interleukin-1beta production from astrocytes, induced by palmitate; implications for Alzheimer’s Disease. Neurobiol. Aging. 2014, 35, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Ma, K. NLRC4 inflammasome activation regulated by TNF-α promotes inflammatory responses in nonalcoholic fatty liver disease. Biochem. Biophys. Res. Commun. 2019, 511, 524–530. [Google Scholar] [CrossRef]
- Bossen, C.; Ingold, K.; Tardivel, A.; Bodmer, J.L.; Gaide, O.; Hertig, S.; Ambrose, C.; Tschopp, J.; Schneider, P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 2006, 281, 13964–13971. [Google Scholar] [CrossRef] [Green Version]
- Kichev, A.; Baburamani, A.A.; Vontell, R.; Gressens, P.; Burkly, L.; Thornton, C.; Hagberg, H. TWEAK Receptor Deficiency Has Opposite Effects on Female and Male Mice Subjected to Neonatal Hypoxia-Ischemia. Front. Neurol. 2018, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Winkles, J.A. The TWEAK-Fn14 cytokine-receptor axis: Discovery, biology and therapeutic targeting. Nat. Rev. Drug Discov. 2008, 7, 411–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yepes, M.; Brown, S.A.; Moore, E.G.; Smith, E.P.; Lawrence, D.A.; Winkles, J.A. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am. J. Pathol. 2005, 166, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Potrovita, I.; Zhang, W.; Burkly, L.; Hahm, K.; Lincecum, J.; Wang, M.Z.; Maurer, M.H.; Rossner, M.; Schneider, A.; Schwaninger, M. Tumor necrosis factor-like weak inducer of apoptosis-induced neurodegeneration. J. Neurosci. 2004, 24, 8237–8244. [Google Scholar] [CrossRef]
- Zhang, X.; Winkles, J.A.; Gongora, M.C.; Polavarapu, R.; Michaelson, J.S.; Hahm, K.; Burkly, L.; Friedman, M.; Li, X.J.; Yepes, M. TWEAK-Fn14 pathway inhibition protects the integrity of the neurovascular unit during cerebral ischemia. J. Cereb. Blood. Flow. Metab. 2007, 27, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Desplat-Jego, S.; Varriale, S.; Creidy, R.; Terra, R.; Bernard, D.; Khrestchatisky, M.; Izui, S.; Chicheportiche, Y.; Boucraut, J. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. J. Neuroimmunol. 2002, 133, 116–123. [Google Scholar] [CrossRef]
- Mustafa, S.; Martin, H.L.; Burkly, L.; Costa, A.; Martins, M.L.; Schwaninger, M.; Teismann, P. The role of TWEAK/Fn14 signaling in the MPTP-model of Parkinson’s disease. Neuroscience 2016, 319, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Saas, P.; Boucraut, J.; Walker, P.R.; Quiquerez, A.L.; Billot, M.; Desplat-Jego, S.; Chicheportiche, Y.; Dietrich, P.Y. TWEAK stimulation of astrocytes and the proinflammatory consequences. Glia 2000, 32, 102–107. [Google Scholar] [CrossRef]
- Gordon, R.; Singh, N.; Lawana, V.; Ghosh, A.; Harischandra, D.S.; Jin, H.; Hogan, C.; Sarkar, S.; Rokad, D.; Panicker, N.; et al. Protein kinase Cdelta upregulation in microglia drives neuroinflammatory responses and dopaminergic neurodegeneration in experimental models of Parkinson’s disease. Neurobiol. Dis. 2016, 93, 96–114. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. Proteolytic activation of proapoptotic kinase protein kinase Cdelta by tumor necrosis factor alpha death receptor signaling in dopaminergic neurons during neuroinflammation. J. Neuroinflamm. 2012, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Kanthasamy, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A. Protein kinase C delta negatively regulates tyrosine hydroxylase activity and dopamine synthesis by enhancing protein phosphatase-2A activity in dopaminergic neurons. J. Neurosci. 2007, 27, 5349–5362. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef]
- Jin, H.; Kanthasamy, A.; Harischandra, D.S.; Kondru, N.; Ghosh, A.; Panicker, N.; Anantharam, V.; Rana, A.; Kanthasamy, A.G. Histone hyperacetylation up-regulates protein kinase Cdelta in dopaminergic neurons to induce cell death: Relevance to epigenetic mechanisms of neurodegeneration in Parkinson disease. J. Biol. Chem. 2014, 289, 34743–34767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Malovic, E.; Plante, B.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Rapid and refined CD11b magnetic isolation of primary microglia with enhanced purity and versatility. JoVE J. Vis. Exp. 2017, 13, e55364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polavarapu, R.; Gongora, M.C.; Winkles, J.A.; Yepes, M. Tumor necrosis factor-like weak inducer of apoptosis increases the permeability of the neurovascular unit through nuclear factor-kappa B pathway activation. J. Neurosci. 2005, 25, 10094–10100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Saminathan, H.; Kanthasamy, A.; Anantharam, V.; Jin, H.; Sondarva, G.; Harischandra, D.S.; Qian, Z.; Rana, A.; Kanthasamy, A.G. The peptidyl-prolyl isomerase Pin1 up-regulation and proapoptotic function in dopaminergic neurons: Relevance to the pathogenesis of Parkinson disease. J. Biol. Chem. 2013, 288, 21955–21971. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 2011, 31, 2035–2051. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free. Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Klemann, C.; Xicoy, H.; Poelmans, G.; Bloem, B.R.; Martens, G.J.M.; Visser, J.E. Physical Exercise Modulates L-DOPA-Regulated Molecular Pathways in the MPTP Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 5639–5657. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Hogan, C.E.; Neal, M.L.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. A simple magnetic separation method for high-yield isolation of pure primary microglia. J. Neurosci. Methods 2011, 194, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Charli, A.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Alterations in mitochondrial dynamics induced by tebufenpyrad and pyridaben in a dopaminergic neuronal cell culture model. Neurotoxicology 2016, 53, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, K.; Lippincott-Schwartz, J. Analysis of mitochondrial dynamics and functions using imaging approaches. Curr. Protoc. Cell Biol. 2010, 393, 1485–1512. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Lawana, V.; Luo, J.; Phong, P.; Abdalla, A.; Palanisamy, B.; Rokad, D.; Sarkar, S.; Jin, H.; Anantharam, V.; et al. Organophosphate pesticide chlorpyrifos impairs STAT1 signaling to induce dopaminergic neurotoxicity: Implications for mitochondria mediated oxidative stress signaling events. Neurobiol. Dis. 2018, 117, 82–113. [Google Scholar] [CrossRef] [PubMed]
- Rai, N.; Upadhyay, A.D.; Goyal, V.; Dwivedi, S.; Dey, A.B.; Dey, S. Sestrin2 as Serum Protein Marker and Potential Therapeutic Target for Parkinson’s Disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [Green Version]
- Madrigal-Matute, J.; Fernandez-Laso, V.; Sastre, C.; Llamas-Granda, P.; Egido, J.; Martin-Ventura, J.L.; Zalba, G.; Blanco-Colio, L.M. TWEAK/Fn14 interaction promotes oxidative stress through NADPH oxidase activation in macrophages. Cardiovasc. Res. 2015, 108, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xiao, S.; Xia, Y. TWEAK/Fn14 Activation Participates in Skin Inflammation. Mediat. Inflamm. 2017, 2017, 6746870. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis. 2017, 3, 30. [Google Scholar] [CrossRef]
- 6Chen, T.; Guo, Z.P.; Li, L.; Li, M.M.; Wang, T.T.; Jia, R.Z.; Cao, N.; Li, J.Y. TWEAK enhances E-selectin and ICAM-1 expression, and may contribute to the development of cutaneous vasculitis. PLoS ONE 2013, 8, e56830. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Peng, H.; Xiang, H.; Guo, L.; Chen, R.; Zhao, S.; Chen, W.; Chen, P.; Lu, H.; Chen, S. TWEAK/Fn14 promotes oxidative stress through AMPK/PGC1alpha/MnSOD signaling pathway in endothelial cells. Mol. Med. Rep. 2018, 17, 1998–2004. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Andres, O.; Suarez-Alvarez, B.; Sanchez-Ramos, C.; Monsalve, M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. The inflammatory cytokine TWEAK decreases PGC-1alpha expression and mitochondrial function in acute kidney injury. Kidney Int. 2016, 89, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.C.; Chang, J.; Chen, W.C. Role of protein kinase C subtypes alpha and delta in the regulation of bradykinin-stimulated phosphoinositide breakdown in astrocytes. Mol. Pharmacol. 1995, 48, 39–47. [Google Scholar] [PubMed]
- Jia, G.; Wang, R.; Yue, Y.; Dai, H. Activation of Protein Kinase Cdelta Contributes to the Induction of Src/EGF Receptor/ERK Signaling in Ammonia-treated Astrocytes. J. Mol. Neurosci. 2020, 10, 1517–1518. [Google Scholar] [CrossRef]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.Y.; Nam, Y.; Kang, H.J.; Cho, H.J.; Kim, J.; Hoe, H.S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflamm. 2018, 15, 271. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, M.; Niidome, T.; Matsuda, S.; Akaike, A.; Kihara, T.; Sugimoto, H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur. J. Neurosci. 2007, 25, 649–658. [Google Scholar] [CrossRef]
- Zhu, P.; Hata, R.; Cao, F.; Gu, F.; Hanakawa, Y.; Hashimoto, K.; Sakanaka, M. Ramified microglial cells promote astrogliogenesis and maintenance of neural stem cells through activation of Stat3 function. FASEB J. 2008, 22, 3866–3877. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Sun, L.; Jia, B.; Lan, X.; Zhu, B.; Wu, Y.; Zheng, J. HIV-1-infected and immune-activated macrophages induce astrocytic differentiation of human cortical neural progenitor cells via the STAT3 pathway. PLoS ONE 2011, 6, e19439. [Google Scholar] [CrossRef]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef]
- Hung, C.-C.; Lee, Y.-H.; Kuo, Y.-M.; Hsu, P.-C.; Tsay, H.-J.; Hsu, Y.-T.; Lee, C.-C.; Liang, J.-J.; Shie, F.-S. Soluble epoxide hydrolase modulates immune responses in activated astrocytes involving regulation of STAT3 activity. J. Neuroinflamm. 2019, 16, 123. [Google Scholar] [CrossRef]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010, 30, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Komuves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.A.; Canna, S.W. The NLRC4 Inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Freemantle, N.; Cleland, J.; Young, P.; Mason, J.; Harrison, J. beta Blockade after myocardial infarction: Systematic review and meta regression analysis. BMJ 1999, 318, 1730–1737. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Langley, M.R.; Harischandra, D.S.; Neal, M.L.; Jin, H.; Anantharam, V.; Joseph, J.; Brenza, T.; Narasimhan, B.; Kanthasamy, A.; et al. Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of Parkinson’s disease. J. Neuroimm. Pharmacol. Off. J. Soc. NeuroImm. Pharmacol. 2016, 11, 259–278. [Google Scholar] [CrossRef] [Green Version]
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H.; et al. Mito-apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in mitopark transgenic mice. Antioxid Redox Signal. 2017, 27, 1048–1066. [Google Scholar] [CrossRef]
- Li, J.; Lv, H.; Che, Y.Q. Upregulated microRNA-31 inhibits oxidative stress-induced neuronal injury through the JAK/STAT3 pathway by binding to PKD1 in mice with ischemic stroke. J. Cell Physiol. 2020, 235, 2414–2428. [Google Scholar] [CrossRef]
- Ng, I.H.; Yeap, Y.Y.; Ong, L.S.; Jans, D.A.; Bogoyevitch, M.A. Oxidative stress impairs multiple regulatory events to drive persistent cytokine-stimulated STAT3 phosphorylation. Biochim. Biophys. Acta 2014, 1843, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.; Dhruv, H.D.; Mathews, I.T.; Inge, L.J.; Tuncali, S.; Hartman, L.K.; Chow, D.; Millard, N.; Yin, H.H.; Kloss, J.; et al. Identification of aurintricarboxylic acid as a selective inhibitor of the TWEAK-Fn14 signaling pathway in glioblastoma cells. Oncotarget 2017, 8, 12234–12246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nature Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.M.; Yin, M.; Zhang, M.H. Cell-based assays for Parkinson’s disease using differentiated human LUHMES cells. Acta Pharmacol. Sinica 2014, 35, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.M.; Han, X.R.; Wen, X.; Wang, S.; Fan, S.H.; Zhuang, J.; Wang, Y.J.; Zhang, Z.F.; Li, M.Q.; Hu, B.; et al. Salidroside Protection Against Oxidative Stress Injury Through the Wnt/beta-Catenin Signaling Pathway in Rats with Parkinson’s Disease. Cell. Physiol. Biochem. 2018, 46, 1793–1806. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Heng, Y.; Mou, Z.; Huang, J.Y.; Yuan, Y.H.; Chen, N.H. Reassessment of subacute MPTP-treated mice as animal model of Parkinson’s disease. Acta Pharmacol. Sin. 2017, 38, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Boulamery, A.; Desplat-Jego, S. Regulation of Neuroinflammation: What Role for the Tumor Necrosis Factor-Like Weak Inducer of Apoptosis/Fn14 Pathway? Front. Immunol. 2017, 8, 1534. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Chen, C.H.; Stock, A.; Doerner, J.; Gulinello, M.; Putterman, C. Intracerebroventricular administration of TNF-like weak inducer of apoptosis induces depression-like behavior and cognitive dysfunction in non-autoimmune mice. Brain Behav. Immun. 2016, 54, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, S.; Chang, S.C.; Lee, J. Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Arch. Pharm. Res. 2019, 42, 416–425. [Google Scholar] [CrossRef]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Kirkley, K.S.; Popichak, K.A.; Hammond, S.L.; Davies, C.; Hunt, L.; Tjalkens, R.B. Genetic suppression of IKK2/NF-kappaB in astrocytes inhibits neuroinflammation and reduces neuronal loss in the MPTP-Probenecid model of Parkinson’s disease. Neurobiol. Dis. 2019, 127, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Rizor, A.; Pajarillo, E.; Johnson, J.; Aschner, M.; Lee, E. Astrocytic oxidative/nitrosative stress contributes to Parkinson’s disease pathogenesis: The Dual Role of Reactive Astrocytes. Antioxidants 2019, 8, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Xu, L.; Zhang, T.; Ren, G.; Yang, Z. Oxidative stress and apoptosis induced by nanosized titanium dioxide in PC12 cells. Toxicology 2010, 267, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Alam, Z.I.; Daniel, S.E.; Lees, A.J.; Marsden, D.C.; Jenner, P.; Halliwell, B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J. Neurochem. 1997, 69, 1326–1329. [Google Scholar] [CrossRef]
- Zhou, C.; Huang, Y.; Przedborski, S. Oxidative stress in Parkinson’s disease: A mechanism of pathogenic and therapeutic significance. Ann. N. Y. Acad. Sci. 2008, 1147, 93–104. [Google Scholar] [CrossRef]
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J. Neurochem. 1997, 69, 1196–1203. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G.; Siedlak, S.L.; Nunomura, A.; Shimohama, S.; Zhang, J.; Montine, T.; Sayre, L.M.; Smith, M.A. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci. Lett. 2002, 319, 25–28. [Google Scholar] [CrossRef]
- Dexter, D.; Carter, C.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Lipid peroxidation as cause of nigral cell death in Parkinson’s disease. Lancet 1986, 2, 639–640. [Google Scholar] [CrossRef]
- Taupin, P. A dual activity of ROS and oxidative stress on adult neurogenesis and Alzheimer’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, M.D.; Sparkman, N.L.; Johnson, R.W. Inhibition of interleukin-6 trans-signaling in the brain facilitates recovery from lipopolysaccharide-induced sickness behavior. J. Neuroinflamm. 2011, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Haim, L.; Ceyzeriat, K.; Carrillo-de Sauvage, M.A.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- McFarland, B.C.; Benveniste, E.N. Reactive astrocytes foster brain metastases via STAT3 signaling. Annals Transl. Med. 2019, 9, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Thangaraj, A.; Guo, M.L.; Hu, G.; Callen, S.; Buch, S. Epigenetic Promoter DNA Methylation of miR-124 Promotes HIV-1 Tat-Mediated Microglial Activation via MECP2-STAT3 Axis. J. Neurosci. 2018, 38, 5367–5383. [Google Scholar] [CrossRef]
- Yu, M.O.; Park, K.J.; Park, D.H.; Chung, Y.G.; Chi, S.G.; Kang, S.H. Reactive oxygen species production has a critical role in hypoxia-induced Stat3 activation and angiogenesis in human glioblastoma. J. Neurooncol. 2015, 125, 55–63. [Google Scholar] [CrossRef]
- Yoon, S.; Woo, S.U.; Kang, J.H.; Kim, K.; Kwon, M.H.; Park, S.; Shin, H.J.; Gwak, H.S.; Chwae, Y.J. STAT3 transcriptional factor activated by reactive oxygen species induces IL6 in starvation-induced autophagy of cancer cells. Autophagy 2010, 6, 1125–1138. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; Di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Denes, A.; Coutts, G.; Lenart, N.; Cruickshank, S.M.; Pelegrin, P.; Skinner, J.; Rothwell, N.; Allan, S.M.; Brough, D. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl. Acad. Sci. USA 2015, 112, 4050–4055. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gamez, M.; Goginashvili, A.; et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef]
- Gong, T.; Jiang, W.; Zhou, R. Control of inflammasome activation by phosphorylation. Trends Biochem. Sci. 2018, 43, 685–699. [Google Scholar] [CrossRef]
- Bednash, J.S.; Mallampalli, R.K. Regulation of inflammasomes by ubiquitination. Cell. Mol. Immunol. 2016, 13, 722–728. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samidurai, M.; Tarale, P.; Janarthanam, C.; Estrada, C.G.; Gordon, R.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.G.; Kanthasamy, A. Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK) Enhances Activation of STAT3/NLRC4 Inflammasome Signaling Axis through PKCδ in Astrocytes: Implications for Parkinson’s Disease. Cells 2020, 9, 1831. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9081831
Samidurai M, Tarale P, Janarthanam C, Estrada CG, Gordon R, Zenitsky G, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A. Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK) Enhances Activation of STAT3/NLRC4 Inflammasome Signaling Axis through PKCδ in Astrocytes: Implications for Parkinson’s Disease. Cells. 2020; 9(8):1831. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9081831
Chicago/Turabian StyleSamidurai, Manikandan, Prashant Tarale, Chelva Janarthanam, Crystal Gomez Estrada, Richard Gordon, Gary Zenitsky, Huajun Jin, Vellareddy Anantharam, Anumantha G. Kanthasamy, and Arthi Kanthasamy. 2020. "Tumor Necrosis Factor-Like Weak Inducer of Apoptosis (TWEAK) Enhances Activation of STAT3/NLRC4 Inflammasome Signaling Axis through PKCδ in Astrocytes: Implications for Parkinson’s Disease" Cells 9, no. 8: 1831. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9081831